

Update README.md
Browse files
README.md
CHANGED
|
@@ -1,3 +1,106 @@
|
|
| 1 |
-
---
|
| 2 |
-
license: apache-2.0
|
| 3 |
-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
---
|
| 2 |
+
license: apache-2.0
|
| 3 |
+
license_link: LICENSE
|
| 4 |
+
tags:
|
| 5 |
+
- chemistry
|
| 6 |
+
- molecular simulations
|
| 7 |
+
- machine learning potentials
|
| 8 |
+
- neural network potentials
|
| 9 |
+
- drug discovery
|
| 10 |
+
---
|
| 11 |
+
|
| 12 |
+
# Acellera AceFF 2.0 - UPCOMING
|
| 13 |
+
|
| 14 |
+
**Organization(s):** Acellera Therapeutics, inc
|
| 15 |
+
**Contact:** info@acellera.com
|
| 16 |
+
**License:** apache 2.0
|
| 17 |
+
|
| 18 |
+
---
|
| 19 |
+
|
| 20 |
+
## Overview
|
| 21 |
+
|
| 22 |
+
Acellera AceFF 2.0 is a next-generation **machine learning interatomic potential (MLIP)** designed for **small molecules**.
|
| 23 |
+
It addresses key limitations of traditional molecular mechanics (MM) force fields and earlier NNP models, including restricted atom types, limited charge support, and computational inefficiencies.
|
| 24 |
+
|
| 25 |
+
The model leverages the TensorNet v2 architecture [1] and the NNP software library TorchMD-Net [2] to provide accurate predictions for diverse drug-like compounds, supporting all key chemical elements and charged molecules.
|
| 26 |
+
Acellera AceFF 2.0 improves the stability of molecular dynamics simulations, supports 2 fs timesteps, and achieves state-of-the-art accuracy with fewer outliers in RBFE predictions.
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
## Description
|
| 30 |
+
|
| 31 |
+
Acellera AceFF 2.0 is the second version of a new family of potentials released by [Acellera](https://www.acellera.com).
|
| 32 |
+
It uses [TensorNet](https://proceedings.neurips.cc/paper_files/paper/2023/file/75c2ec5f98d7b2f50ad68033d2c07086-Paper-Conference.pdf) 2-layers trained
|
| 33 |
+
on Acellera's internal proprietary dataset of molecular forces and energies using the wB97M-V/def2-tzvppd level of theory and VV10 dispersion corrections.
|
| 34 |
+
|
| 35 |
+
The training set was built on [PubChem](https://ftp.ncbi.nlm.nih.gov/pubchem/Compound/CURRENT-Full/SDF).
|
| 36 |
+
We extracted the SMILES and generated molecules, filtering out molecules larger than 30 atoms.
|
| 37 |
+
We kept only molecules with the elements H, B, C, N, O, F, Si, P, S, Cl, Br, and I.
|
| 38 |
+
|
| 39 |
+
---
|
| 40 |
+
|
| 41 |
+
## Bechmarks
|
| 42 |
+
|
| 43 |
+
### Wiggle150
|
| 44 |
+
The table shows the results on the [Wiggle150](https://pubs.acs.org/doi/10.1021/acs.jctc.5c00015) benchmark. We include AIMNet2 and ANI-2x for comparison.
|
| 45 |
+
|
| 46 |
+
| **Method** | **MAE (kcal/mol)** | **RMSE (kcal/mol)** |
|
| 47 |
+
|------------|--------------------|---------------------|
|
| 48 |
+
| AceFF-2.0 | | |
|
| 49 |
+
| AceFF-1.1 | 2.51 | 3.18 |
|
| 50 |
+
| AceFF-1.0 | 2.73 | 3.32 |
|
| 51 |
+
| AIMNet2 | 2.39 | 3.13 |
|
| 52 |
+
| ANI-2X | 4.41 | 5.41 |
|
| 53 |
+
|
| 54 |
+
*Performance of MLIPs on Wiggle150 benchmark*
|
| 55 |
+
|
| 56 |
+
### Schrodinger ligands test set
|
| 57 |
+
We create our own hold-out test set by labelling 650 ligands from the [Schrodinger public binding free energy benchmark](https://github.com/schrodinger/public_binding_free_energy_benchmark) (Jacs, Merk, and charge_annhil sets) with AceFF's DFT level of theory. We evaluate the Force MAE of the AceFF predictions.
|
| 58 |
+
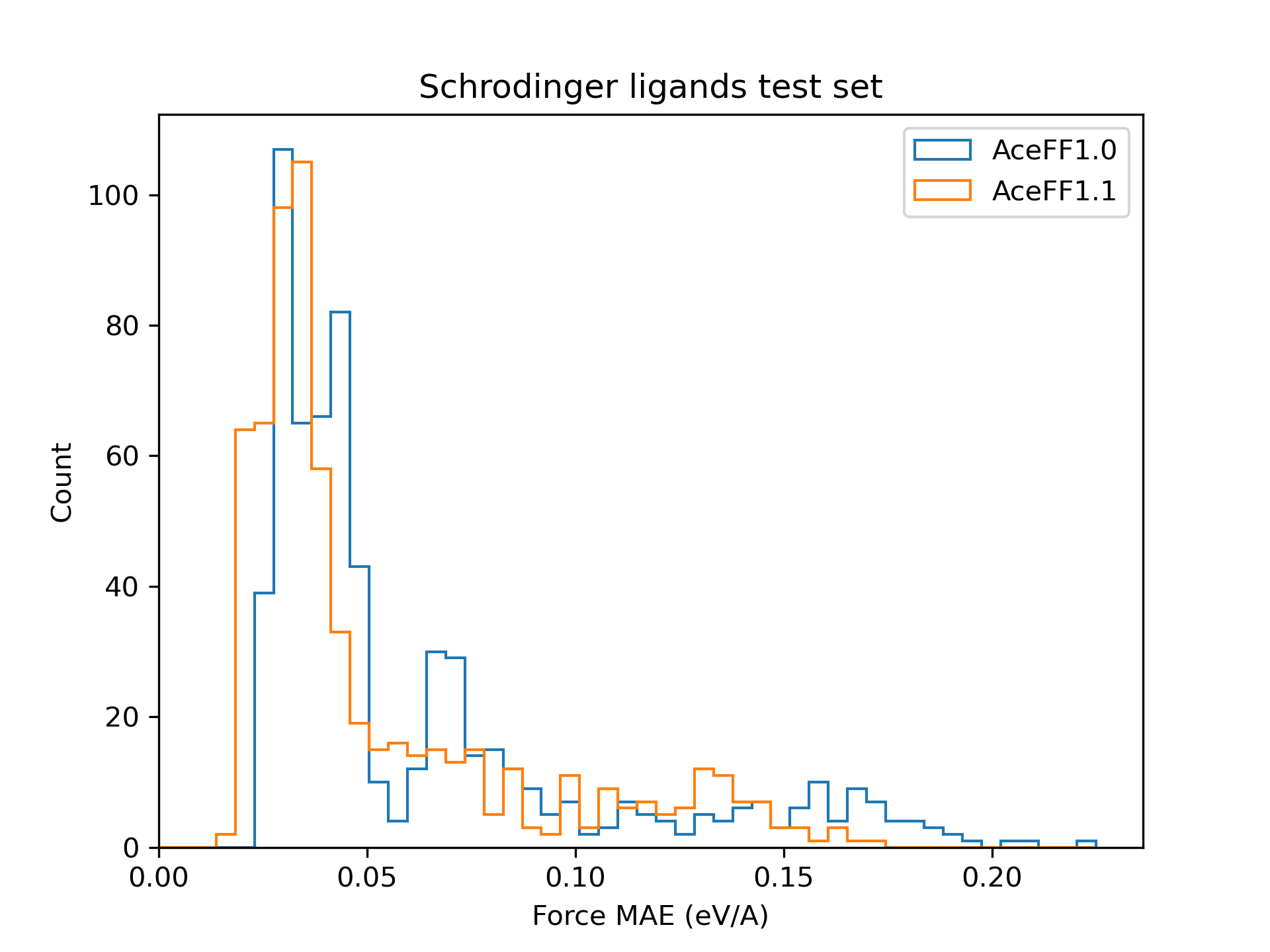
|
| 59 |
+
|
| 60 |
+
---
|
| 61 |
+
|
| 62 |
+
## Key Features
|
| 63 |
+
|
| 64 |
+
- **Broad Applicability:** Supports diverse drug-like molecules, including charged species and rare chemical groups.
|
| 65 |
+
- **High Accuracy:** Benchmark-tested on the JACS dataset, demonstrating performance comparable to or better than MM-based methods (e.g., GAFF2, FEP+).
|
| 66 |
+
- **Improved Stability:** Enables a 2 fs timestep for NNP/MM simulations, significantly reducing computational costs.
|
| 67 |
+
- **Integration-Friendly:** Available for RBFE calculations via [HTMD](https://github.com/acellera/htmd).
|
| 68 |
+
- **Open Science:** The model and all benchmarking data are accessible on GitHub for not-for-profit usage.
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
---
|
| 72 |
+
|
| 73 |
+
## Usage
|
| 74 |
+
|
| 75 |
+
1. [Example notebooks](https://github.com/Acellera/aceff_examples) are available in **Google Colab**, demonstrating the use of Acellera AceFF with OpenMM and ASE.
|
| 76 |
+
|
| 77 |
+
- Single point calculation with ASE [](https://colab.research.google.com/github/Acellera/aceff_examples/blob/main/notebooks/aceff_single_point_calculation.ipynb)
|
| 78 |
+
- ML molecular dynamics of a small molecule with OpenMM [](https://colab.research.google.com/github/Acellera/aceff_examples/blob/main/notebooks/aceff_MD_example.ipynb)
|
| 79 |
+
- MM/ML protein-ligand simulations with OpenMM [](https://colab.research.google.com/github/Acellera/aceff_examples/blob/main/notebooks/aceff_protein_ligand.ipynb)
|
| 80 |
+
2. Run ML potential molecular simulations of a small molecule using ACEMD with this [tutorial](https://software.acellera.com/acemd/nnp.html), e.g., to minimize.
|
| 81 |
+
3. For a tutorial on running mixed protein-ligand simulations, refer to [NNP/MM in ACEMD](https://software.acellera.com/acemd/nnpmm.html).
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
---
|
| 85 |
+
|
| 86 |
+
## Applications
|
| 87 |
+
|
| 88 |
+
- **Drug Discovery:** Optimizing lead compounds in hit-to-lead and lead optimization stages using free energy methods.
|
| 89 |
+
- **Binding Free Energy Calculations:** Accurate and efficient RBFE predictions for diverse molecular systems.
|
| 90 |
+
- **Molecular dynamics:** Capturing higher-body terms than traditional MM force fields, Acellera AceFF can be used for structure minimization and dynamics of small molecules.
|
| 91 |
+
|
| 92 |
+
## Limitations
|
| 93 |
+
|
| 94 |
+
- **Small molecules only**: Acellera AceFF-2.0 is trained on specifically curated and extended PubChem data. However, proteins, water, etc are not part of the dataset right now.
|
| 95 |
+
- **Time step**: Use time steps of 2fs to run dynamics with hydrogen mass repartitioning.
|
| 96 |
+
- **Only -2,-1,0,1,2 charges**: For simplicity, we have trained only on these types of charged molecules.
|
| 97 |
+
|
| 98 |
+
---
|
| 99 |
+
|
| 100 |
+
## References
|
| 101 |
+
|
| 102 |
+
[1] Simeon, Guillem, and Gianni De Fabritiis, Tensornet: Cartesian tensor representations for efficient learning of molecular potentials, Advances in Neural Information Processing Systems 36 (2024), https://arxiv.org/abs/2306.06482
|
| 103 |
+
|
| 104 |
+
[2] Raul P. Pelaez, Guillem Simeon, Raimondas Galvelis, Antonio Mirarchi, Peter Eastman, Stefan Doerr, Philipp Thölke, Thomas E. Markland, Gianni De Fabritiis, TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations, J. Chem. Theory Comput. 2024, 20, 10, 4076–4087, https://arxiv.org/abs/2402.17660
|
| 105 |
+
|
| 106 |
+
[3] Francesc Sabanés Zariquiey, Stephen E. Farr, Stefan Doerr, Gianni De Fabritiis, QuantumBind-RBFE: Accurate Relative Binding Free Energy Calculations Using Neural Network Potentials, https://arxiv.org/abs/2501.01811 (2025).
|