
ESM Protein Function
Collection
Protein language models fine-tuned for function prediction using the Gene Ontology. • 12 items • Updated • 1

An Evolutionary-scale Model (ESM) for protein function prediction from amino acid sequences using the Gene Ontology (GO). Based on the ESM Cambrian Transformer architecture, pre-trained on UniRef, MGnify, and the Joint Genome Institute's database and fine-tuned on the AmiGO Boost protein function dataset, this protein language model predicts the GO subgraph for a particular protein sequence - giving you insight into the molecular function, biological process, and location of the activity inside the cell.
Sequence-to-function prediction — Predicts Molecular Function, Biological Process, and Cellular Component ontologies directly from raw amino acid sequences, eliminating the need for homology searches, structural data, or multiple sequence alignments.
Hierarchy-aware GO subgraph reconstruction — Outputs a full GO directed acyclic graph (DAG) ensuring predictions respect the ontology structure rather than treating each term as an independent binary label.
Efficient inference at scale — Supports weight quantization and quantization-aware training (QAT), enabling memory-efficient, high-throughput screening of large sequence datasets without accuracy loss.
"The Gene Ontology (GO) is a concept hierarchy that describes the biological function of genes and gene products at different levels of abstraction (Ashburner et al., 2000). It is a good model to describe the multi-faceted nature of protein function."
"GO is a directed acyclic graph. The nodes in this graph are functional descriptors (terms or classes) connected by relational ties between them (is_a, part_of, etc.). For example, terms 'protein binding activity' and 'binding activity' are related by an is_a relationship; however, the edge in the graph is often reversed to point from binding towards protein binding. This graph contains three subgraphs (subontologies): Molecular Function (MF), Biological Process (BP), and Cellular Component (CC), defined by their root nodes. Biologically, each subgraph represent a different aspect of the protein's function: what it does on a molecular level (MF), which biological processes it participates in (BP) and where in the cell it is located (CC)."
From CAFA 5 Protein Function Prediction
The code repository can be found at https://github.com/andrewdalpino/ESMC-Protein-Function.
The following pretrained models are available on HuggingFace Hub and require the esmc-protein-function library version 1.x.x for inference. All V1 models have been optimized with quantization-aware post-training.
| Name | Embedding Dimensions | Encoder Layers | Context Length | Total Parameters |
|---|---|---|---|---|
| andrewdalpino/ESMC-Protein-Function-V1-300M | 960 | 30 | 2048 | 397M |
| andrewdalpino/ESMC-Protein-Function-V1-600M | 1152 | 36 | 2048 | 661M |
The following pretrained models are available on HuggingFace Hub and require the esmc_function_classifier library version 0.1.x for inference.
| Name | Embedding Dimensions | Encoder Layers | Context Length | Total Parameters |
|---|---|---|---|---|
| andrewdalpino/ESMC-Protein-Function-V0-300M | 960 | 30 | 2048 | 361M |
| andrewdalpino/ESMC-Protein-Function-V0-300M-QAT | 960 | 30 | 2048 | 361M |
| andrewdalpino/ESMC-Protein-Function-V0-600M | 1152 | 36 | 2048 | 644M |
| andrewdalpino/ESMC-Protein-Function-V0-600M-QAT | 1152 | 36 | 2048 | 644M |
First, install the esmc-protein-function package using pip. I recommend using a virtual environment such as venv to keep dependencies compartmentalized.
pip install esmc-protein-function
Then, we'll load the model weights from HuggingFace Hub by calling the from_pretrained() method. We'll also need the ESM tokenizer from the esm library.
from esmc_protein_function.model import ESMCProteinFunction
model_name = "andrewdalpino/ESMC-Protein-Function-V1-300M"
device = "cpu" # Can be "cpu", "cuda", "mps", etc.
model = ESMCProteinFunction.from_pretrained(model_name)
model = model.to(device)
For faster inference and lower memory usage you can cast the weights of the model to a lower bitdepth than the default float32.
import torch
# Cast weights to float16.
model = model.to(dtype=torch.float16)
# Or quantize the weights to Int8.
model.quantize_weights(group_size=64)
We'll also need the ESM tokenizer from the esm library to tokenize the amino acid sequences.
from esm.tokenization import EsmSequenceTokenizer
tokenizer = EsmSequenceTokenizer()
In this example we'll predict the Gene Ontology terms for all apsects of a protein sequence. The return values are dicts with GO terms as keys and predicted probabilities as values.
sequence = "MPPKGHKKTADGDFRPVNSAGNTIQAKQKYSIDDLLYPKSTIKNLAKETLPDDAIISKDALTAIQRAATLFVSYMASHGNASAEAGGRKKIT"
top_p = 0.5
out = tokenizer(sequence, max_length=2048, truncation=True)
x = torch.tensor(out["input_ids"], dtype=torch.int32)
# Add the batch dimension for a single sequence.
x = x.unsqueeze(0)
x = x.to(device)
mf_terms, bp_terms, cc_terms = model.predict_all_terms(x, top_p=top_p)
print(mf_terms[0])
print(bp_terms[0])
print(cc_terms[0])
You can also query individual apsects of the Gene Ontology using the predict_mf_terms(), predict_bp_terms(), and predict_cc_terms() methods like in the example below.
mf_terms = model.predict_mf_terms(x, top_p=top_p)
bp_terms = model.predict_bp_terms(x, top_p=top_p)
cc_terms = model.predict_cc_terms(x, top_p=top_p)
You can also output the GO networkx subgraph for a given sequence. You'll need an up-to-date Gene Ontology database that you can import using the obonet package. The database can be downloaded at https://geneontology.org/docs/download-ontology/.
pip install obonet
Then, load the GO DAG and call the predict_all_subgraphs() method like in the example below.
import networkx as nx
import obonet
go_db_path = "./dataset/go-basic.obo"
graph = obonet.read_obo(go_db_path)
model.load_gene_ontology(graph)
mf_subgraphs, bp_subgraphs, cc_subgraphs = model.predict_all_subgraphs(x, top_p=top_p)
# Render the subgraphs ...
See the predict_subgraphs.py file in the repository for an example of how to render the GO DAGs using Plotly.
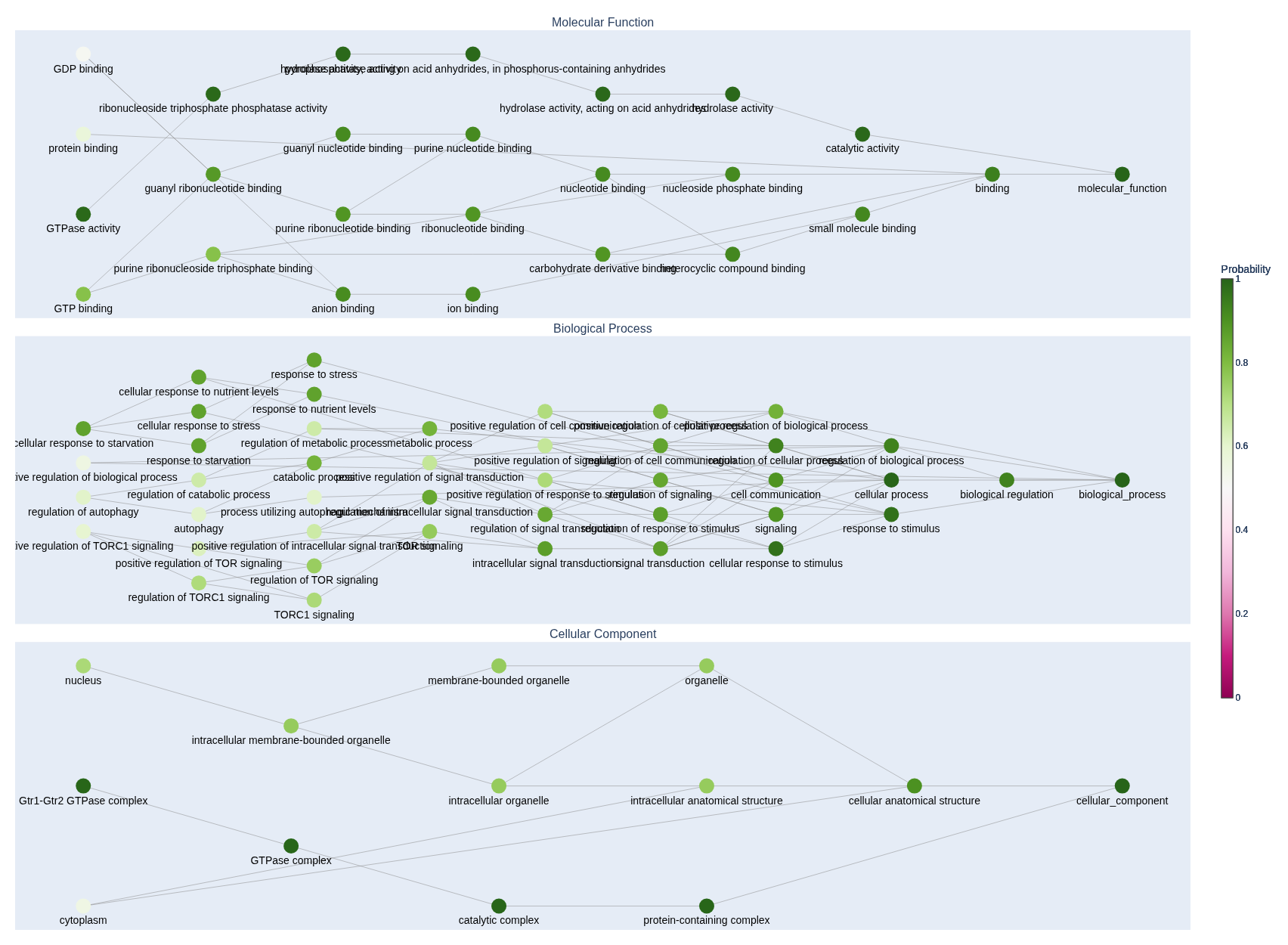
You can also ouput the subgraphs for individual apsects of the GO using the predict_mf_subgraphs(), predict_bp_subgraphs(), and predict_cc_subgraphs() methods like in the example below.
mf_subgraphs = model.predict_mf_subgraphs(x, top_p=top_p)
bp_subgraphs = model.predict_bp_subgraphs(x, top_p=top_p)
cc_subgraphs = model.predict_cc_subgraphs(x, top_p=top_p)
An ESMC Protein Function model inference server can be found at https://github.com/andrewdalpino/ESMC-GO-Inference-Server.
- T. Hayes, et al. Simulating 500 million years of evolution with a language model, 2024.
- M. Ashburner, et al. Gene Ontology: tool for the unification of biology, 2000.
- Z. Lin, et al. A Structured Self-attentive Sentence Embedding, ICLR 2017.