Buckets:

Title: Learning Inter-Atomic Potentials without Explicit Equivariance
URL Source: https://arxiv.org/html/2510.00027
Markdown Content: Ahmed A.Elhag
Computer Science
University of Oxford
&Arun Raja 1 1 footnotemark: 1
Statistics
University of Oxford
&Alex Morehead 1 1 footnotemark: 1
NERSC
Lawrence Berkeley National Laboratory
&Samuel M.Blau
Energy Technologies Area
Lawrence Berkeley National Laboratory
&Garrett M.Morris
Statistics
University of Oxford
&Michael M.Bronstein
Computer Science
University of Oxford
Abstract
Accurate and scalable machine-learned inter-atomic potentials (MLIPs) are essential for molecular simulations ranging from drug discovery to new material design. Current state-of-the-art models enforce roto-translational symmetries through equivariant neural network architectures, a hard-wired inductive bias that can often lead to reduced flexibility, computational efficiency, and scalability. In this work, we introduce TransIP: Trans former-based I nter-Atomic P otentials, a novel training paradigm for interatomic potentials achieving symmetry compliance without explicit architectural constraints. Our approach guides a generic non-equivariant Transformer-based model to learn SO(3)\mathrm{SO}(3)-equivariance by optimizing its representations in the embedding space. Trained on the recent Open Molecules (OMol25) collection, a large and diverse molecular dataset built specifically for MLIPs and covering different types of molecules (including small organics, biomolecular fragments, and electrolyte-like species), TransIP effectively learns symmetry in its latent space, providing low equivariance error. Further, compared to a data augmentation baseline, TransIP achieves 40% to 60% improvement in performance across varying OMol25 dataset sizes. More broadly, our work shows that learned equivariance can be a powerful and efficient alternative to augmentation-based MLIP models.
1 Introduction
Atomistic simulations are a fundamental task in chemistry and materials science (Zhang et al., 2018; Deringer et al., 2019), with Density Functional Theory (DFT) serving as a basis for accurately calculating interatomic forces and energies. However, the utility of DFT is severely restricted by its computational costs, which typically scale cubically with system size, rendering large-scale or long-timescale simulations intractable. This has motivated machine-learned interatomic potentials (MLIPs) to overcome this limitation by learning the potential energy surface from data, offering orders-of-magnitude speed-ups compared to DFT calculations (Noé et al., 2020; Batzner et al., 2022; Batatia et al., 2022; Jacobs et al., 2025; Leimeroth et al., 2025).
Equivariant neural networks have become a central paradigm for MLIPs due to their ability to encode the three-dimensional structure of molecular graphs (Anderson et al., 2019; Thölke & Fabritiis, 2022; Liao et al., 2024a; Fu et al., 2025). These architectures are designed to explicitly respect roto-translational symmetries (SE(3) equivariance) by construction, often employing compute-intensive mechanisms like spherical harmonics or equivariant message passing (Fuchs et al., 2020; Passaro & Zitnick, 2023a; Liao & Smidt, 2023; Maruf et al., 2025). However, due to the design difficulties and limited expressive power of these architectures (Joshi et al., 2023; Cen et al., 2024), a recent trend in predictive and generative modeling is to use unconstrained models when enough data is available (Wang et al., 2024; Abramson et al., 2024; Zhang et al., 2025; Joshi et al., 2025).
In this paper, we introduce TransIP (Trans former-based I nteratomic P otentials), a training paradigm that achieves molecular symmetry for interatomic potentials without imposing architectural SO(3)\mathrm{SO}(3) constraints. TransIP steers a standard transformer toward SO(3)\mathrm{SO}(3) equivariance via an additional contrastive objective, allowing the model to retain the scalability and hardware efficiency of attention mechanisms while learning symmetry from data.
Figure 1: TransIP: Transformer-based Interatomic Potentials.
Our contributions are as follows:
- •We propose an MLIP training pipeline with a general transformer-based model to obtain SO(3)\mathrm{SO}(3) equivariance through training rather than hard-wired equivariant layers.
- •We introduce an architecture-agnostic contrastive loss function that promotes SO(3)\mathrm{SO}(3) equivariance in the embedding space of an unconstrained model. By aligning latent features across SO(3)\mathrm{SO}(3) transformations in the model’s backbone, we show that TransIP scales better across different datasets and model sizes compared to traditional data augmentation techniques.
- •On a diverse molecular benchmark, Open Molecules 25 (Levine et al., 2025) (that includes small organics, biomolecular fragments, electrolyte-like species), we show that TransIP outperforms data augmentation techniques often by a large margin, which might provide an alternative for the future of augmentation-based MLIP models.
2 Symmetry in Embedding Space
2.1 Problem Formulation
Molecular representations. Let ℳ\mathcal{M} denote the space of molecular configurations. Each molecule m∈ℳ m\in\mathcal{M} is represented by atomic features 𝐱=(𝐫,𝐳,q,s)\mathbf{x}=(\mathbf{r},\mathbf{z},q,s), where 𝐫∈ℝ|m|×3\mathbf{r}\in\mathbb{R}^{|m|\times 3} are atomic coordinates, 𝐳∈ℕ|m|\mathbf{z}\in\mathbb{N}^{|m|} are atomic numbers, q∈ℤ q\in\mathbb{Z} is the total molecular charge, and s∈ℕ s\in\mathbb{N} is the spin multiplicity, with |m||m| denoting the number of atoms in molecule m m.
Our goal is to learn an embedding function f θ:ℳ→ℝ d f_{\theta}:\mathcal{M}\rightarrow\mathbb{R}^{d} that maps molecular configurations to a d d-dimensional latent space, and a prediction function g φ:ℝ d→ℝ g_{\varphi}:\mathbb{R}^{d}\rightarrow\mathbb{R} that acts in the embedding space ℝ d\mathbb{R}^{d} and outputs molecular properties (e.g., energy). Both f θ f_{\theta} and g φ g_{\varphi} are neural networks parameterized by θ\theta and φ\varphi, respectively.
Symmetry groups. We define a symmetry group G G that acts on a set 𝒳\mathcal{X} as a group of bijective functions from 𝒳\mathcal{X} to itself, and the group operation is function composition. We say a function f f is equivariant w.r.t. the group G G if for every transformation g∈G g\in G and every input x∈X x\in X,
f(ϕ(g)(x))=ρ(g)(f(x))f(\phi(g)(x))=\rho(g)(f(x))(1)
The group representations ϕ\phi and ρ\rho specify how we apply the elements of the group G G on input and output data. As a concrete case, we can define G G as a rotation group SO(3)\mathrm{SO}(3) over molecular configurations ℳ\mathcal{M}, with g∈SO(3)g\in\mathrm{SO}(3) representing an element of G G that acts on a molecule m m by rotating the coordinates of each atom in 3D space. Formally, for a molecule m=(𝐫,𝐳,q,s)m=(\mathbf{r},\mathbf{z},q,s) with coordinates 𝐫=(𝐫 1,…,𝐫|m|)\mathbf{r}=(\mathbf{r}{1},\ldots,\mathbf{r}{|m|}), 𝐫 i∈ℝ 3\mathbf{r}_{i}\in\mathbb{R}^{3}, the input action rotates each atom:
(ϕ(g)m)=((R𝐫 1,…,R𝐫|m|),𝐳,q,s).\bigl(\phi(g),m\bigr)=\bigl((R\mathbf{r}{1},\ldots,R\mathbf{r}{|m|}),,\mathbf{z},,q,,s\bigr).
Here R R is a 3×3 3\times 3 rotation matrix (orthogonal with det R=1\det R=1); 𝐳,q,s\mathbf{z},q,s are unchanged. An associated output representation rotates vector-valued quantities—e.g., for forces 𝐅=(𝐅 1,…,𝐅|m|)\mathbf{F}=(\mathbf{F}{1},\ldots,\mathbf{F}{|m|}), ρ(g)𝐅=(R𝐅 1,…,R𝐅|m|)\rho(g)\mathbf{F}=(R\mathbf{F}{1},\ldots,R\mathbf{F}{|m|})—while scalar outputs such as energies remain invariant, ρ(g)E=E\rho(g)E=E.
2.2 Implicit Equivariance in Embedding Space
We seek an embedding function f f that behaves equivariantly with respect to the symmetry group G G, meaning there exists a transformation ρ(g):ℝ d→ℝ d\rho(g):\mathbb{R}^{d}\rightarrow\mathbb{R}^{d} such that:
f(ϕ(g)(m))=ρ(g)(f(m))∀g∈G,m∈ℳ f(\phi(g)(m))=\rho(g)(f(m))\quad\forall g\in G,m\in\mathcal{M}(2)
Common approaches enforce equivariance constraints through specialized architectures. Instead, we want the embedding function f f to learn symmetry without equivariance constraints. However, with G G being the rotation group SO(3)\mathrm{SO}(3) on ℳ\mathcal{M} and the output of f f being a high-dimensional vector, there is no direct representation of ρ(g)\rho(g) to act in the space of ℝ d\mathbb{R}^{d}. Thus, rather than specifying ρ(g)\rho(g) analytically, we propose to learn the group transformation on an embedding vector in ℝ d\mathbb{R}^{d} using a neural network 𝒯 τ:SO(3)×ℝ d→ℝ d\mathcal{T}_{\tau}:\text{SO}(3)\times\mathbb{R}^{d}\rightarrow\mathbb{R}^{d} parameterized by τ\tau. 𝒯\mathcal{T} can be understood as a non-linear function that learns the group action implicitly on a latent vector, by providing the group representation on the input data.
3 Learning Inter-Atomic Potentials without Explicit Equivariance
In this section, we introduce our training framework: TransIP (Transformer-based Inter-atomic Potentials), a new approach that achieves SO(3)\mathrm{SO}(3)-equivariance through learned transformations in an embedding space without explicit equivariance constraints. Our method, illustrated in Figure1, consists of three key components: (i) an unconstrained Transformer backbone that processes molecular configurations, (ii) a learned transformation network that performs group actions in the embedding space, and (iii) a contrastive objective that enforces latent equivariance (equiv.) during training.
3.1 TransIP: Transformer-based Interatomic Potentials
Atom as tokens. We model each molecule as a variable-length sequence of tokens, where each token represents an atom. Unlike conventional graph neural networks that construct edges based on distance cutoffs or neighbours’ atoms, we process all atoms within a molecule through self-attention, bounded by a maximum context length N ctx N_{\text{ctx}}. For batch processing, we use padding masks to prevent cross-molecule attention, ensuring each molecule is processed independently.
In addition, we apply rotary position embeddings (RoPE) (Su et al., 2023) to the queries 𝐪 i∈ℝ d/h\mathbf{q}{i}\in\mathbb{R}^{d/h} and keys 𝐤 j∈ℝ d/h\mathbf{k}{j}\in\mathbb{R}^{d/h} of each attention head, where i,j i,j denote the sequence positions of atoms within a molecule, d d is the model dimension, and h h is the number of attention heads. The attention weights are computed as:
𝐪i\displaystyle\tilde{\mathbf{q}}_{i}=RoPE(𝐪 i,i),𝐤j=RoPE(𝐤 j,j)\displaystyle=\mathrm{RoPE}(\mathbf{q}{i},i),\quad\tilde{\mathbf{k}}{j}=\mathrm{RoPE}(\mathbf{k}{j},j)
α ij\displaystyle\alpha{ij}=softmax(𝐪i⊤𝐤j d/h+m ij)\displaystyle=\mathrm{softmax}\left(\frac{\tilde{\mathbf{q}}{i}^{\top}\tilde{\mathbf{k}}{j}}{\sqrt{d/h}}+m_{ij}\right)
where RoPE(⋅,⋅)\mathrm{RoPE}(\cdot,\cdot) is the rotary position encoding operator, and m ij∈{0,−∞}m_{ij}\in{0,-\infty} is the attention mask that blocks padding tokens and enforces within-molecule attention. This approach eliminates the need for explicit distance cutoffs while maintaining flexibility in modeling molecular interactions.
Transformer Backbone. We implement the embedding function f θ:ℳ→ℝ d f_{\theta}:\mathcal{M}!\to!\mathbb{R}^{d} as a Transformer encoder that processes atom-level tokens. Each atom i i is initialized with a token representation:
𝐡 i(0)=κ 𝐳(z i)⊕κ 𝐫(𝐫 i)\mathbf{h}{i}^{(0)}=\kappa{\mathbf{z}}(z_{i})\oplus\kappa_{\mathbf{r}}(\mathbf{r}_{i})
where κ 𝐳:ℕ→ℝ d\kappa_{\mathbf{z}}:\mathbb{N}!\to!\mathbb{R}^{d} and κ 𝐫:ℝ 3→ℝ d\kappa_{\mathbf{r}}:\mathbb{R}^{3}!\to!\mathbb{R}^{d} are learnable MLPs that embed atomic numbers and centered coordinates (with 𝐫 i←𝐫 i−1|m|∑j 𝐫 j\mathbf{r}{i}\leftarrow\mathbf{r}{i}-\tfrac{1}{|m|}\sum_{j}\mathbf{r}{j}), and ⊕\oplus denotes concatenation. These tokens are processed through L L Transformer layers with masked self-attention within each molecule, producing final per-atom embeddings 𝐇=[𝐡 1,…,𝐡|m|]⊤∈ℝ|m|×d\mathbf{H}=[\mathbf{h}{1},\dots,\mathbf{h}_{|m|}]^{\top}\in\mathbb{R}^{|m|\times d}.
Global Molecular Properties. Following Levine et al. (2025), we incorporate global molecular properties (total charge q q and spin multiplicity s s of a molecule m m) through learnable embeddings, and form a graph-level bias:
𝐜(q,s)=κ chg(q)+κ spin(s)∈ℝ d\mathbf{c}(q,s)=\kappa_{\mathrm{chg}}(q)+\kappa_{\mathrm{spin}}(s)\in\mathbb{R}^{d}
where κ chg\kappa_{\mathrm{chg}} and κ spin\kappa_{\mathrm{spin}} are learnable embedding functions for charge and spin, respectively. This global bias is broadcast-added at each Transformer layer: 𝐇(ℓ)←𝐇(ℓ)+𝟏𝐜(q,s)⊤\mathbf{H}^{(\ell)}\leftarrow\mathbf{H}^{(\ell)}+\mathbf{1}\mathbf{c}(q,s)^{\top}.
Energy and Force Predictions. For molecular property prediction, we employ a permutation-invariant aggregator a:ℝ|m|×d→ℝ d a:\mathbb{R}^{|m|\times d}!\to!\mathbb{R}^{d} followed by an energy prediction head g φ:ℝ d→ℝ g_{\varphi}:\mathbb{R}^{d}!\to!\mathbb{R}:
E φ(m)=g φ(a(𝐇))E_{\varphi}(m)=g_{\varphi}(a(\mathbf{H}))
Forces are computed as conservative gradients of the energy with respect to atomic positions:
𝐅(m)=−∇𝐫 E φ(m)∈ℝ|m|×3\mathbf{F}(m)=-\nabla_{\mathbf{r}}E_{\varphi}(m)\in\mathbb{R}^{|m|\times 3}
3.2 Learned Latent Equivariance
Transformation Network. We propose a transformation network 𝒯 τ:SO(3)×ℝ d→ℝ d\mathcal{T}{\tau}:\mathrm{SO}(3)\times\mathbb{R}^{d}\rightarrow\mathbb{R}^{d} that learns how group actions (e.g., rotations) act on molecular embeddings. We implement 𝒯 τ\mathcal{T}{\tau} as a multilayer perceptron that takes as input the group representation in the input domain ϕ(g)\phi(g) and the molecular embedding f(m)f(m). Formally,
𝒯 τ(ϕ(g),f(m))=MLP τ([ϕ(g),f(m)])\mathcal{T}{\tau}(\phi(g),f(m))=\text{MLP}{\tau}([\phi(g),f(m)])
where [⋅,⋅][\cdot,\cdot] denotes concatenation and MLP τ is a multilayer perceptron with parameters τ\tau.
Contrastive Objective for Latent Equivariance: To learn the molecular symmetry without architectural constraints, we define our latent equivariance loss as:
ℒ leq(ϕ(g),m,f,𝒯)=‖f(ϕ(g)(m))−𝒯 τ(ϕ(g),f(m))‖2\mathcal{L}{\textit{leq}}(\phi(g),m,f,\mathcal{T})=|f(\phi(g)(m))-\mathcal{T}{\tau}(\phi(g),f(m))|^{2}(3)
This loss encourages the embedding function f f to behave equivariantly with respect to the symmetry group G G, as mediated by the transformation network 𝒯 τ\mathcal{T}_{\tau}. During training, we sample a molecule m m from the dataset and a rotation element g g uniformly from SO(3)\mathrm{SO}(3) and minimize the expected latent loss:
min𝔼 m∼ℳ,g∼SO(3)[ℒ leq(ϕ(g),m,f,𝒯)]\min\mathbb{E}_{m\sim\mathcal{M},g\sim\mathrm{SO}(3)}\mathcal{L}_{\textit{leq}}(\phi(g),m,f,\mathcal{T})
3.3 Training Objective
Our training objective combines three complementary losses for accurate prediction of energy and forces as well as implicitly learning molecular symmetry.
Prediction Losses. For energy and force predictions, we use:
ℒ E\displaystyle\mathcal{L}{E}=1|m||E φ(m)−E⋆|(per-atom mean absolute error (MAE))\displaystyle=\tfrac{1}{|m|}|E{\varphi}(m)-E^{\star}|\quad\text{(per-atom mean absolute error (MAE))}(5) ℒ F\displaystyle\mathcal{L}{F}=1 3|m|‖𝐅(m)−𝐅⋆‖F 2(per-molecule mean squared error (MSE))\displaystyle=\tfrac{1}{3|m|}|\mathbf{F}(m)-\mathbf{F}^{\star}|{F}^{2}\quad\text{(per-molecule mean squared error (MSE))}(6)
where E⋆E^{\star} and 𝐅⋆\mathbf{F}^{\star} are ground-truth energies and forces, and ∥⋅∥F|\cdot|_{F} denotes the Frobenius norm. For energies, we use referenced targets as described by Levine et al. (2025).
Combined Objective. Training combines three weighted terms: (i) the latent equivariance target ℒ leq\mathcal{L}{\textit{leq}} defined in Eq.3; (ii) energy loss ℒ E\mathcal{L}{E}; and (iii) force loss ℒ F\mathcal{L}_{F}. The total objective is
ℒ total=λ Eℒ E+λ Fℒ F+λ leqℒ leq\mathcal{L}{\mathrm{total}};=;\lambda{E}\mathcal{L}{E};+;\lambda{F}\mathcal{L}{F};+;\lambda{\textit{leq}},\mathcal{L}_{\textit{leq}}(7)
where λ E\lambda_{E}, λ F\lambda_{F}, and λ leq\lambda_{\textit{leq}} are hyperparameters for each loss. The optimal hyperparameters are given in Table 3 of Appendix A.
4 Related Work
ML Interatomic Potentials. Using machine learning (ML) methods to predict energies and forces of different molecular systems and materials has been an active area of research (Schütt et al., 2017; Chmiela et al., 2022; Musaelian et al., 2023; Liao et al., 2024b; Yang et al., 2025; Yuan et al., 2025). Due to the intricate 3D structures of atomistic systems, equivariant message-passing neural networks have been an essential backbone in this domain. For example, Gasteiger et al. (2020); Klicpera et al. (2021) introduced equivariant directional message passing between pairs of atoms with a spherical harmonics representation. In contrast, Batzner et al. (2022) developed equivariant convolution with tensor-products and Batatia et al. (2022) built higher-order messages with equivariant graph neural networks (Satorras et al., 2021). Additionally, Passaro & Zitnick (2023b) reduced the computational complexity of SO(3) convolution and replaced it with SO(2) convolutions, which have been used as a backbone for MLIPs (Fu et al., 2025). More recently, Rhodes et al. (2025) presented Orb-v3 models with improved computational efficiency, built on Graph Network Simulators (Sanchez-Gonzalez et al., 2020).
Unconstrained ML models. While current-state-of-the-art MLIP models primarily rely on equivariant GNNs, unconstrained models are actively used in other domains. For example, integrating data augmentation via image transformations has been used in different vision tasks, from classification (Inoue, 2018; Dosovitskiy et al., 2021; Rahat et al., 2024) to segmentation (Negassi et al., 2022; Yu et al., 2023). For geometric data, the use of unconstrained models and diffusion Transformers (without explicit equivariance constraints) has been a recent trend in generative tasks, e.g., AlphaFold 3 for biomolecular structure prediction (Abramson et al., 2024) as well as molecular conformation and materials generation (Wang et al., 2024; Zhang et al., 2025; Joshi et al., 2025). In contrast, several works have been introduced to overcome the limitations of strictly equivariant GNNs by enforcing symmetry via frame averaging over geometric inputs (Puny et al., 2022; Duval et al., 2023; Lin et al., 2024; Huang et al., 2024; Dym et al., 2024); learning canonicalization functions that map inputs to a canonical orientation before prediction (Kaba et al., 2022; Baker et al., 2024; Ma et al., 2024; Lippmann et al., 2025); or learning equivariance through data augmentation with molecule-specific graph-based architectures (Qu & Krishnapriyan, 2024; Mazitov et al., 2025). However, in this work, we demonstrate that an unconstrained general-purpose Transformer model can serve as a backbone for MLIPs, which replaces graph-based inductive biases with a scalable latent equivariance objective that implicitly learns equivariant features without explicit equivariance constraints.
5 Experimental Setup
Dataset. We train and evaluate our proposed method TransIP on the Open Molecules 2025 (OMol25) collection (Levine et al., 2025), a large-scale molecular DFT dataset for ML interatomic potentials. OMol25 covers 83 atomic elements and diverse chemistries (such as neutral organics, biomolecules, electrolytes, and metal complexes). Following Levine et al. (2025), we use the official 4M training split (3,986,754) and the out-of-distribution composition validation split Val-Comp (2,762,021). Val-Comp consists of molecules gathered from various datasets and domains, such as biomolecules, neutral organics, and metal complexes.
Model Configurations. We evaluate TransIP across three model scales: Small (14M parameters), Medium (85M parameters), and Large (302M parameters). All models use MLP-based coordinate embeddings and RoPE positional encodings. The transformation network 𝒯 τ\mathcal{T}_{\tau} is a 2-layer MLP with GELU activations and 2d 2\mathrm{d} hidden dimension.
Training Setup. Using the standardized fairchem Python package (Shuaibi et al., 2025), we train TransIP on the OMol25 dataset using an AdamW optimizer with learning rate 5×10−4 5\times 10^{-4}, weight decay 10−3 10^{-3}, and gradient norm clipping at 200. We use a cosine learning rate schedule with linear warmup over the first 1% of training, followed by cosine decay down to 1% of the initial lr. The loss weights are set to λ E=5\lambda_{E}=5 for energies and λ F=15\lambda_{F}=15 for forces. For the latent equivariance objective λ leq\lambda_{\textit{leq}}, we sweep the values in {1,5,10,100}{1,5,10,100} and selected λ leq=5\lambda_{\textit{leq}}=5 based on validation performance.
Scalability Experiments. We conduct three sets of experiments to assess TransIP’s scaling behavior:
- •Data scaling: We train the Small (14M parameter) model on three dataset sizes (1M, 2M, 4M molecules) for 5 epochs using 8 NVIDIA 80GB GPUs, comparing TransIP with learned equivariance against an unconstrained Transformer version with SO(3)\mathrm{SO(3)} data augmentation (TransAug).
- •Model size scaling. We compare TransIP and TransAug with different model sizes (Small/Medium/Large) trained on the same number of samples from the OMol25 4M dataset and report the evaluation metrics as a function of the processed number of atoms per second.
- •Extended training: We train TransIP (Small) on the OMol25 4M dataset for 40 epochs using 64 NVIDIA 80GB GPUs to evaluate its performance against standardized equivariant baselines.
Baselines. We compare TransIP against: (i) an unconstrained TransIP variant trained with SO(3) rotation augmentation to assess the impact of learned latent equivariance versus data augmentations, and (ii) state-of-the-art equivariant models on OMol25: eSCN(Fu et al., 2025) in small/medium configurations with both direct and energy-conserving force variants as well as GemNet-OC(Gasteiger et al., 2022).
Evaluation metrics. Following the OMol25 official benchmark, we report: Force MAE (eV/Å), Force cosine similarity, Energy per atom MAE (eV/atom), and Total energy MAE (eV). Detailed metric definitions are provided in AppendixA.4.
6 Results and Discussion
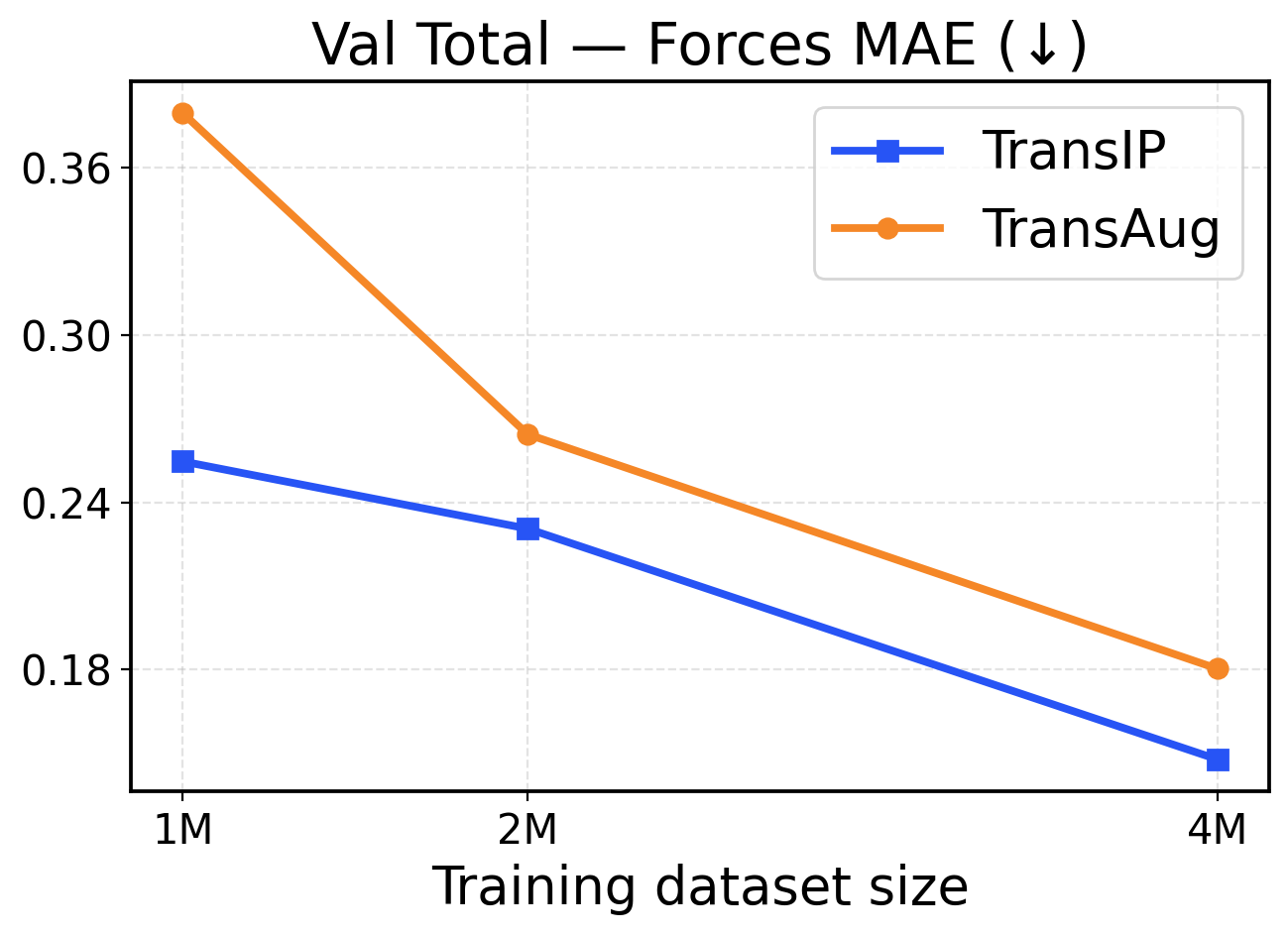

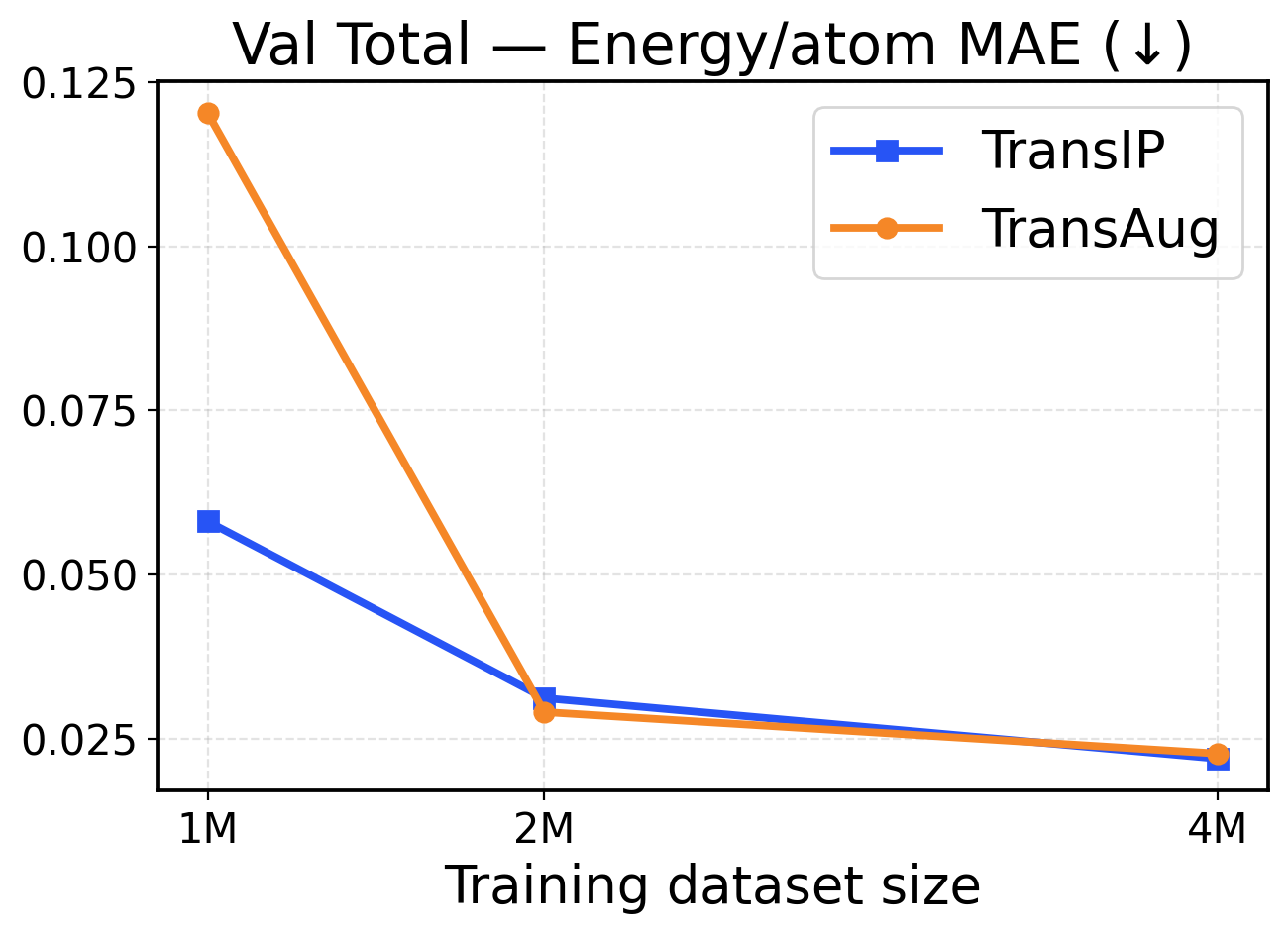
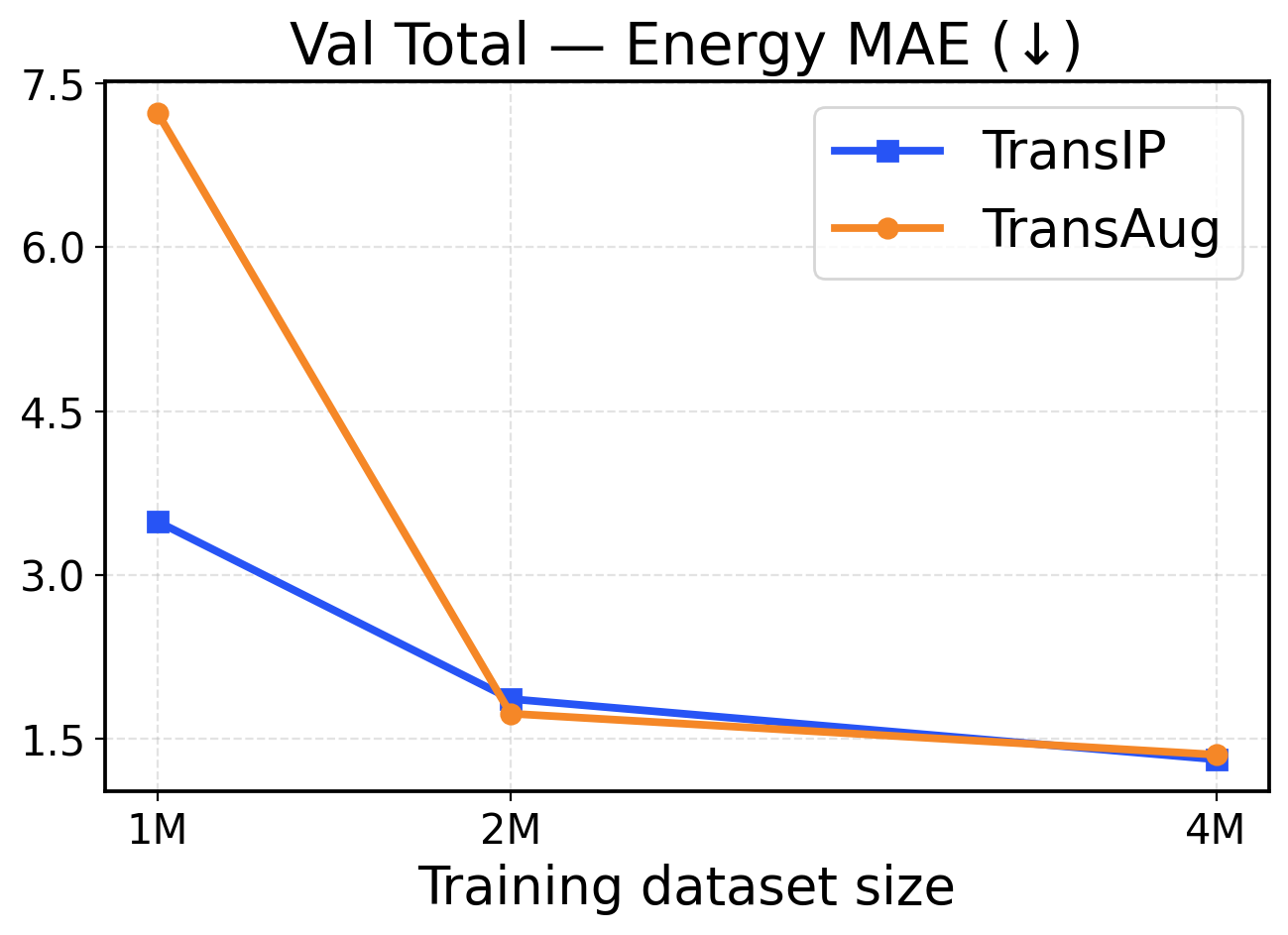
Figure 2: Val-Comp performance across different dataset sizes (1M / 2M / 4M): The top row presents force metrics, while the bottom row reports energy metrics.
6.1 Scaling data size
To assess how performance scales with different training dataset sizes, we compare our latent equivariance-based model (TransIP) against an unconstrained baseline that uses SO(3)\mathrm{SO(3)} data augmentation (TransAug). Both models use a (small) 14M parameter Transformer architecture. Given our tight compute budget, we train on 1M, 2M, and 4M OMol25 molecules for 5 epochs and report validation (Val-Comp) results.
Performance in a limited data regime. Figure 2 shows that TransIP delivers large gains when trained on 1 1 M samples and outperforms TransAug across all evaluation metrics with a large margin. The learned latent equivariance objective provides substantial improvements in force MAE (0.255 0.255 eV/Å vs 0.6 0.6 eV/Å MAE) and directional consistency (0.7 0.7 vs 0.44 0.44 force cosine similarity). Energy predictions also benefit from the latent equivariance objective, with TransIP achieving 0.0581 0.0581 eV/atom compared to TransAug’s 0.1203 0.1203 eV/atom. These results suggest that learning equivariance in a latent space is a more effective scheme to incorporate molecular symmetry than data augmentation, particularly when training data is limited.
Performance in a larger data regime. As we scale to 2 2 M and 4 4 M molecules, both models (TransIP and TransAug) improve across the evaluation metrics. However, on larger datasets, TransIP still achieves better force MAEs and cosine similarity metrics compared to TransAug. This might indicate that the learned transformation network can successfully capture the geometric relationships necessary for accurate force predictions. Notably, energy prediction performance converges between the two at larger data scales, with both methods achieving comparable per-atom MAE values. This convergence suggests that while learned equivariance provides crucial benefits for force-related metrics in all data regimes, its advantages for energy prediction become less pronounced as the model can learn invariant energy representations from sufficient augmented data.
6.2 Learned latent equivariance
We investigate how learned equivariance affects the embedding space in relation to validation performance as the data scale increases. Figure3 plots each metric against latent equiv. error for TransIP (Small) trained for 5 epochs on 1M, 2M, and 4M molecules (see Table2 for a detailed definition of each model configuration).
Lower latent equivariance error leads to better accuracy. We found that the learned equiv. error serves as a strong predictor of model performance. Across all metrics, we observe a clear monotonic trend: lower equiv. error is associated with better performance (Figure3). However, energy and force predictions respond differently to improvements in equivariance. Energy predictions show near-linear scaling with equiv. error, indicating that energy accuracy is directly limited by equivariance quality. This strong coupling aligns with energies being scalar invariants that depend primarily on learning correct symmetry-preserving features. In contrast, force predictions exhibit a two-regime behavior: initial improvements in equivariance (1M→2M) yield modest force improvements, while further tightening of equivariance (2M→4M) produces disproportionate gains. This might indicate that forces require both accurate equivariant features and sufficient data diversity to learn the energy landscape’s geometry.
These results demonstrate that implicitly learning equivariance through our learned transformation network provides an efficient inductive bias, accelerating learning. The 48% reduction in equiv. error from 1M to 4M training examples translates to 40-60% performance improvements, being more efficient than what would be expected from data scaling alone.
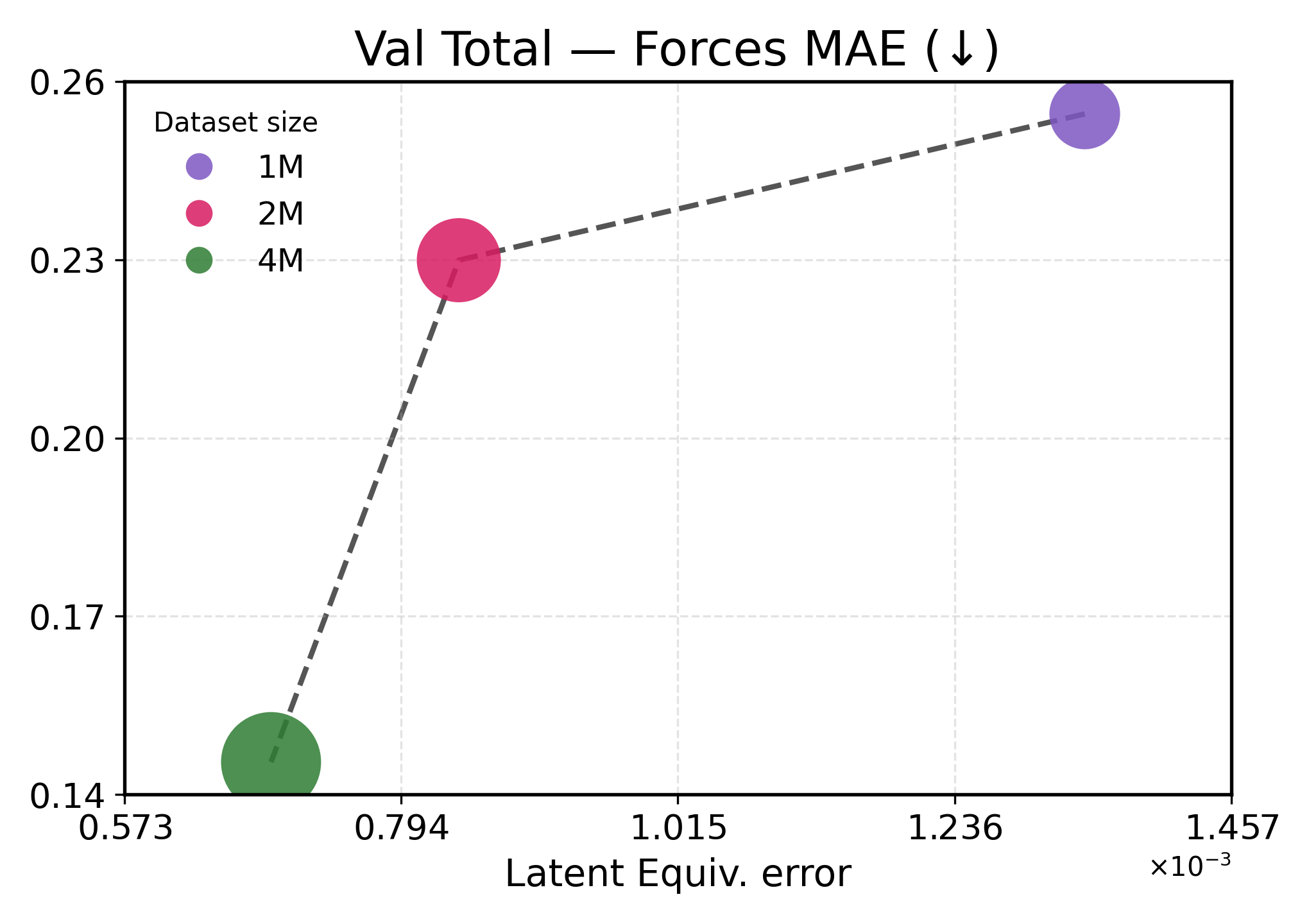
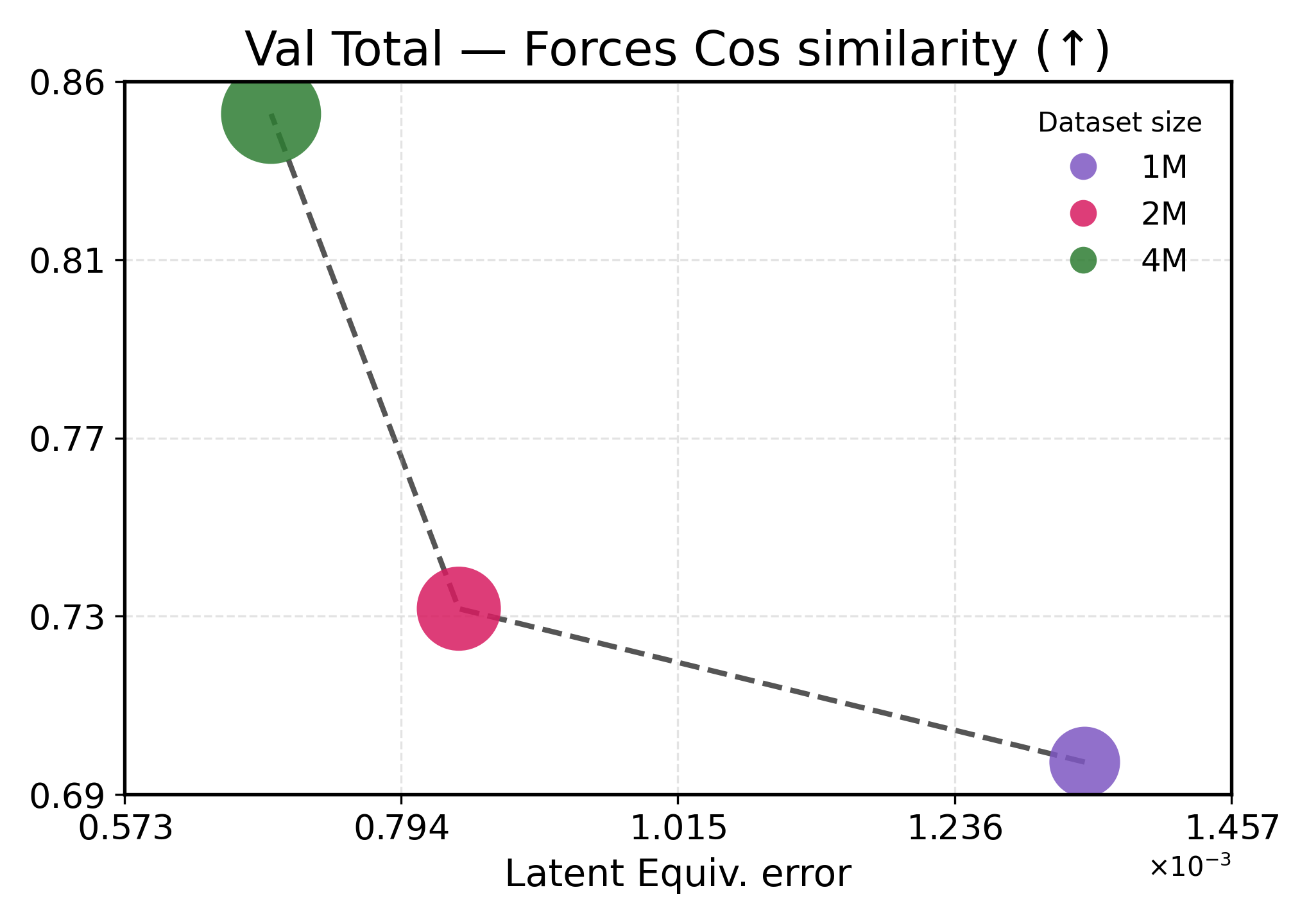
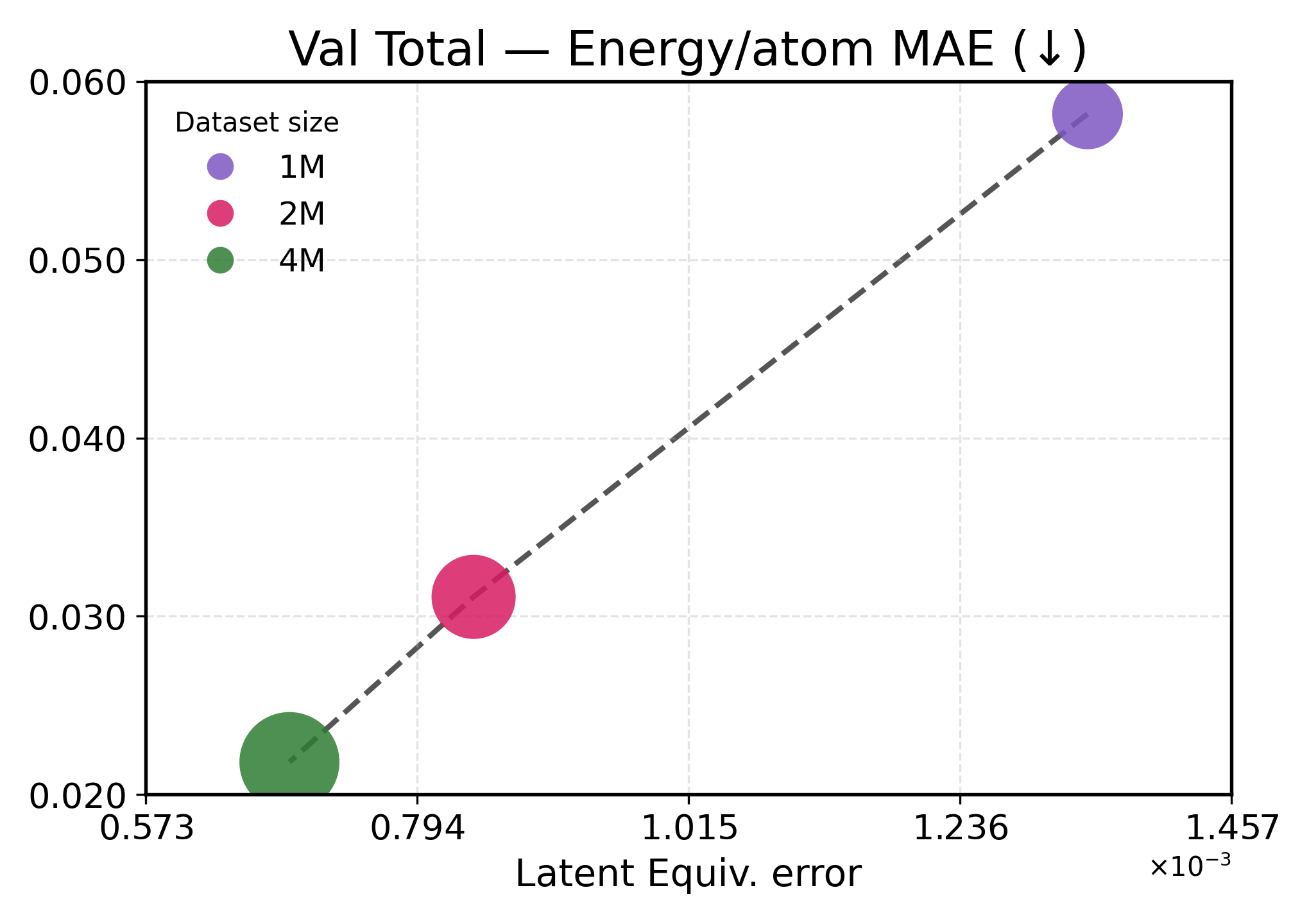
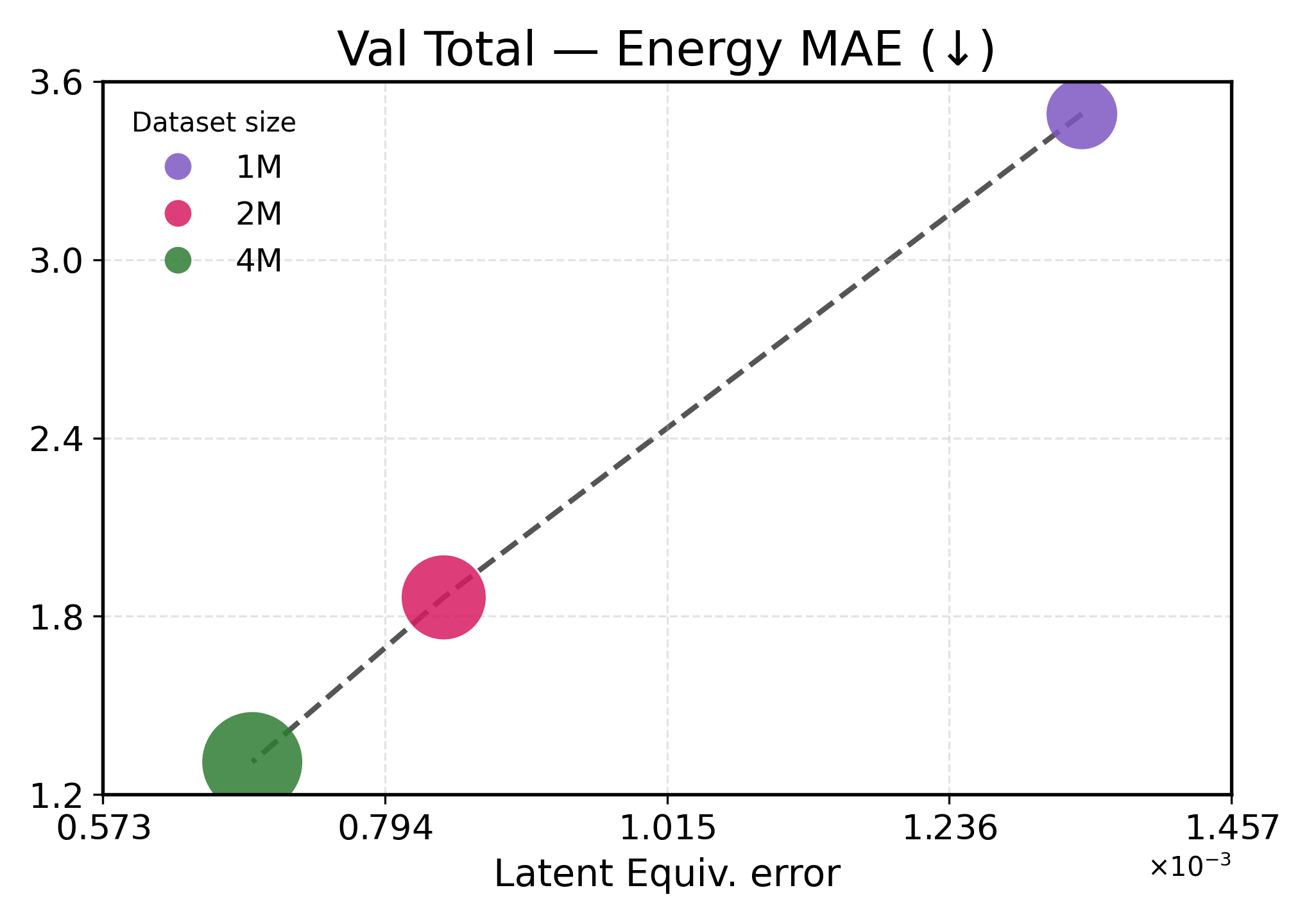
Figure 3: Latent equivariance (embedding) error versus validation performance. The top row reports force metrics, while the bottom row presents energy metrics.
Learning equivariance leads to faster inference. To measure the inference efficiency of our method, we compare TransIP and TransAug with different model sizes (Small/Medium/Large) trained on 4M samples and report the evaluation metrics as a function of the processed number of atoms per second. However, due to limited compute, we compare models under a fixed training budget (i.e., with the same number of samples), which is 10k, 25k, and 100k steps for our Small, Medium, and Large models, respectively.
From the results in Figure 4, we see that TransIP scales smoothly with parameter count despite limited training: As model size grows, performance improves across all metrics. In contrast, TransAug exhibits poorer scaling—larger models perform worse than smaller ones, with the Large model configuration yielding the lowest performance. This might indicate that augmentation alone does not provide a sufficiently informative and stable inductive bias for large-capacity models trained for molecular force field prediction.
6.3 Architectural equivariance versus learned equivariance
Table1 compares the energy and force prediction performance of TransIP (Small) against TransAug (Small) as well as several well-known equivariant baselines for the OMol 2M Val-Comp evaluation dataset. The results of this comparison demonstrate that TransIP outperforms TransAug (trained for 5 epochs) in all but one evaluation metric, particularly differentiating itself in terms of force prediction. We further report the performance of TransIP trained for 40 epochs and, due to limited compute, we are currently training the model for a full 80 epochs (for fair comparison to each equivariant baseline). Results with TransIP after 40 training epochs suggest steady improvement is likely to be observed during the remainder of the model’s training epochs.
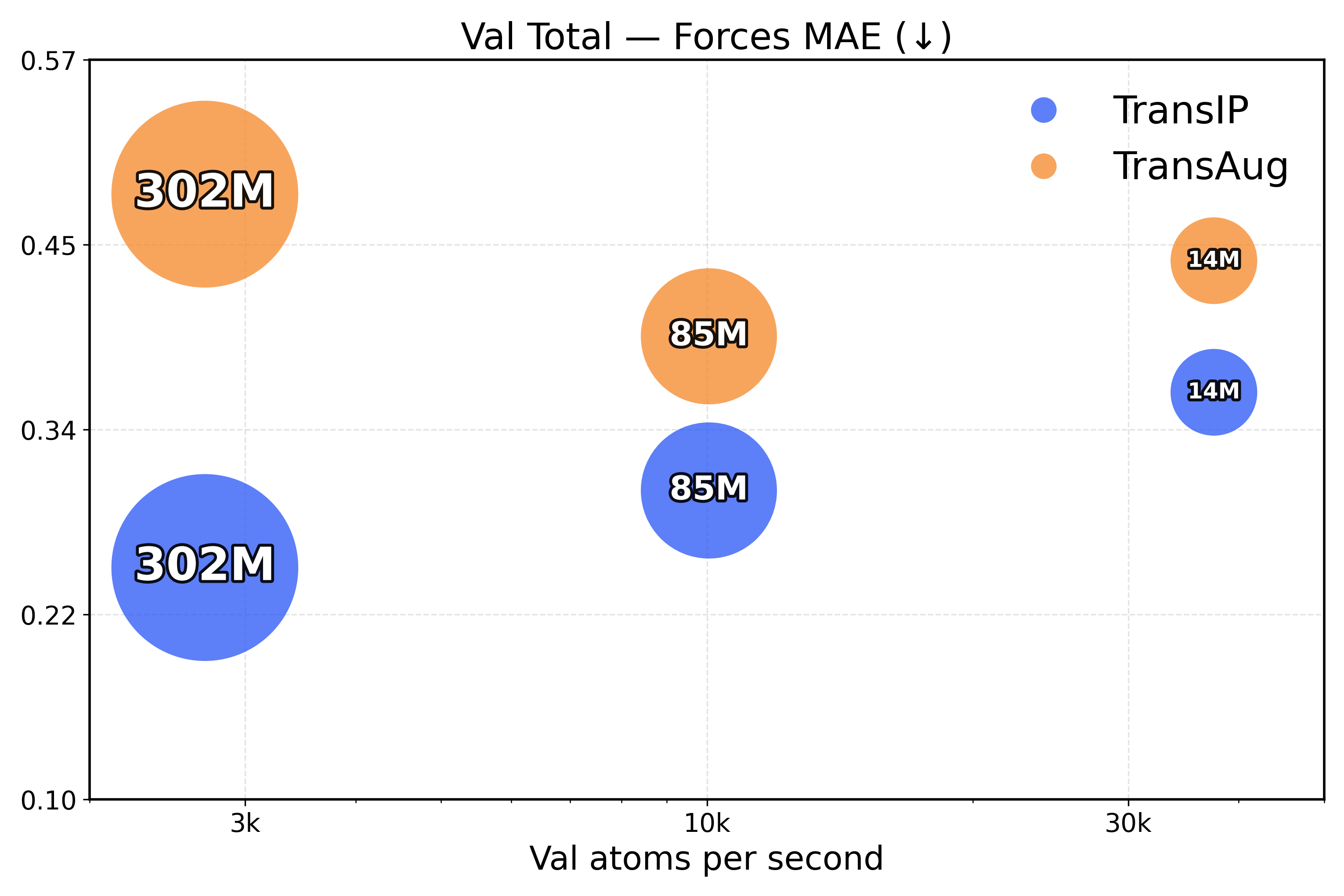
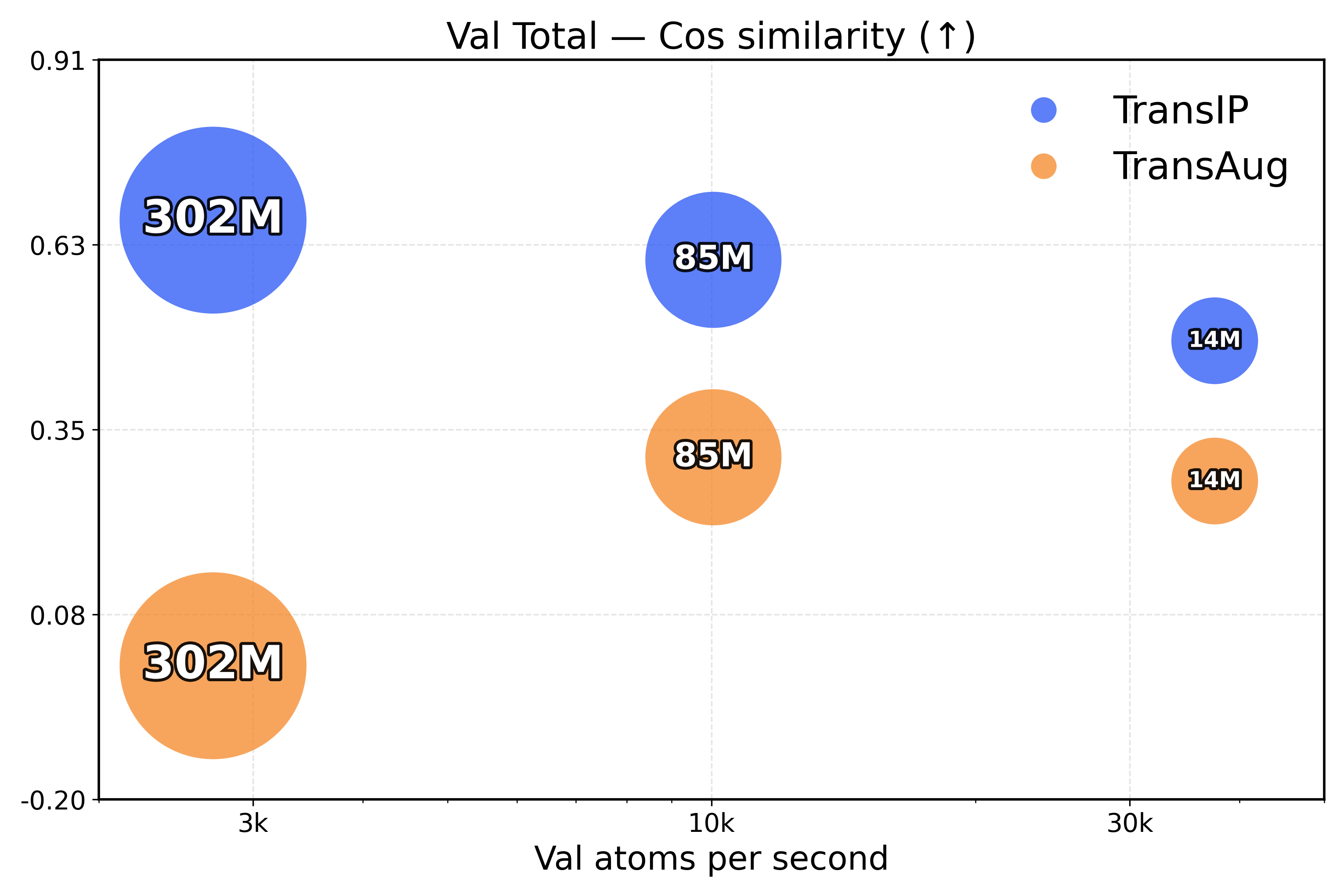

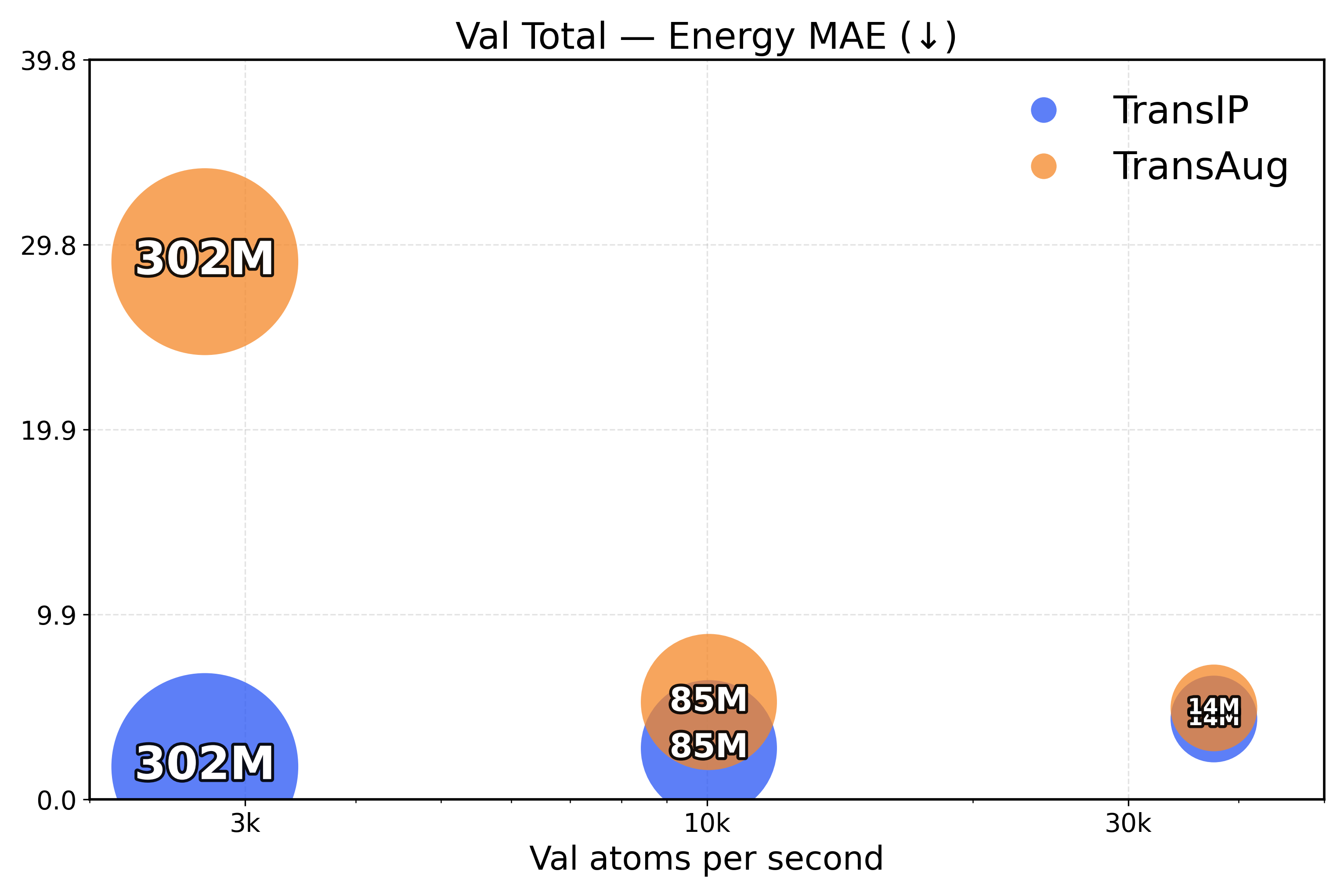
Figure 4: Validation total inference trade-off (atoms/s versus performance). The top row presents force metrics, while the bottom row represents energy metrics.
Table 1: Comprehensive Val-Comp energy and force MAE results.
7 Conclusion
In this work, we introduced TransIP for modeling interatomic potentials with a modern Transformer-based architecture and a scalable latent equivariance objective. Empirical results across a variety of chemical systems as well as model and dataset scales suggest that TransIP’s latent equivariance objective enables better performance scaling than popular data augmentation-based alternatives to learning geometric equivariance. Further, we find that improvements in learning latent equivariance are strongly related to improved modeling of interatomic potentials, suggesting a complementary nature between the two prediction objectives. With sufficient compute, future work could involve studying the performance of TransIP in larger data, modeling, and runtime regimes in addition to the behavior of TransIP in a context amenable to the double-descent phenomenon (Power et al., 2022).
While equivariant models for molecular machine learning have recently gained much research interest, with the large amount of data being generated and the need for larger model sizes, it is also important that models used for interatomic potentials be highly scalable. Through our work, we have shown that the generic Transformer is capable of modeling molecules accurately but is also able to learn equivariance effectively through our novel latent objective, all while being highly scalable. By making our code openly available to the research community, we hope that our work inspires future research that explores ways to leverage the simpler and more scalable Transformer architecture to better model equivariant molecular properties through learned equivariance.
Acknowledgments
This research is partially supported by EPSRC Turing AI World-Leading Research Fellowship No. EP/X040062/1, EPSRC AI Hub on Mathematical Foundations of Intelligence: An “Erlangen Programme” for AI No. EP/Y028872/1. Further, this research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy (DOE) User Facility, using NERSC award DDR-ERCAP 0034574 awarded to AM. S.M.B. acknowledges support from the Center for High Precision Patterning Science (CHiPPS), an Energy Frontier Research Center funded by the U.S. DOE, Office of Science, Basic Energy Sciences (BES). AR’s PhD is supported by the Agency for Science Technology and Research and the SABS R3 CDT program via the Engineering and Physical Sciences Research Council. AR also received compute resources from the DSO National Laboratories - AI Singapore (AISG) programme and the Lawrence Livermore National Laboratory. We would like to thank them for their resources, which played a significant role in this research. We would also like to thank Santiago Vargas, Chaitanya Joshi, and Chen Lin for their fruitful discussions.
References
- Abramson et al. (2024) Jacob Abramson, Jane Adler, John Dunger, et al. Accurate structure prediction of biomolecular interactions with alphafold 3. Nature, 630:493–500, 2024. doi: 10.1038/s41586-024-07487-w. URL https://doi.org/10.1038/s41586-024-07487-w.
- Anderson et al. (2019) Brandon Anderson, Truong Son Hy, and Risi Kondor. Cormorant: Covariant molecular neural networks. In Advances in Neural Information Processing Systems, volume 32. Curran Associates, Inc., 2019. URL https://proceedings.neurips.cc/paper_files/paper/2019/file/03573b32b2746e6e8ca98b9123f2249b-Paper.pdf.
- Baker et al. (2024) Justin Baker, Shih-Hsin Wang, Tommaso de Fernex, and Bao Wang. An explicit frame construction for normalizing 3d point clouds. In Forty-first International Conference on Machine Learning, 2024. URL https://openreview.net/forum?id=SZ0JnRxi0x.
- Batatia et al. (2022) Ilyes Batatia, David Peter Kovacs, Gregor N.C. Simm, Christoph Ortner, and Gabor Csanyi. MACE: Higher order equivariant message passing neural networks for fast and accurate force fields. In Alice H. Oh, Alekh Agarwal, Danielle Belgrave, and Kyunghyun Cho (eds.), Advances in Neural Information Processing Systems, 2022. URL https://openreview.net/forum?id=YPpSngE-ZU.
- Batzner et al. (2022) Simon Batzner, Albert Musaelian, Lixin Sun, et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nature Communications, 13:2453, 2022. doi: 10.1038/s41467-022-29939-5. URL https://doi.org/10.1038/s41467-022-29939-5.
- Cen et al. (2024) Jiacheng Cen, Anyi Li, Ning Lin, Yuxiang Ren, Zihe Wang, and Wenbing Huang. Are high-degree representations really unnecessary in equivariant graph neural networks? In The Thirty-eighth Annual Conference on Neural Information Processing Systems, 2024. URL https://openreview.net/forum?id=M0ncNVuGYN.
- Chmiela et al. (2022) Stefan Chmiela, Valentin Vassilev-Galindo, Oliver T. Unke, Adil Kabylda, Huziel E. Sauceda, Alexandre Tkatchenko, and Klaus-Robert Müller. Accurate global machine learning force fields for molecules with hundreds of atoms, 2022. URL https://arxiv.org/abs/2209.14865.
- Deringer et al. (2019) Volker L. Deringer, Miguel A. Caro, and Gábor Csányi. Machine learning interatomic potentials as emerging tools for materials science. Advanced Materials, 2019.
- Dosovitskiy et al. (2021) Alexey Dosovitskiy, Lucas Beyer, Alexander Kolesnikov, Dirk Weissenborn, Xiaohua Zhai, Thomas Unterthiner, Mostafa Dehghani, Matthias Minderer, Georg Heigold, Sylvain Gelly, Jakob Uszkoreit, and Neil Houlsby. An image is worth 16x16 words: Transformers for image recognition at scale. In International Conference on Learning Representations, 2021. URL https://openreview.net/forum?id=YicbFdNTTy.
- Duval et al. (2023) Alexandre Agm Duval, Victor Schmidt, Alex Hernández-García, Santiago Miret, Fragkiskos D. Malliaros, Yoshua Bengio, and David Rolnick. FAENet: Frame averaging equivariant GNN for materials modeling. In Proceedings of the 40th International Conference on Machine Learning, 2023. URL https://proceedings.mlr.press/v202/duval23a.html.
- Dym et al. (2024) Nadav Dym, Hannah Lawrence, and Jonathan W. Siegel. Equivariant frames and the impossibility of continuous canonicalization. In ICML, 2024. URL https://openreview.net/forum?id=4iy0q0carb.
- Fu et al. (2025) Xiang Fu, Brandon M Wood, Luis Barroso-Luque, Daniel S. Levine, Meng Gao, Misko Dzamba, and C.Lawrence Zitnick. Learning smooth and expressive interatomic potentials for physical property prediction. In Forty-second International Conference on Machine Learning, 2025. URL https://openreview.net/forum?id=R0PBjxIbgm.
- Fuchs et al. (2020) Fabian B. Fuchs, Daniel E. Worrall, Volker Fischer, and Max Welling. Se(3)-transformers: 3d roto-translation equivariant attention networks. In Advances in Neural Information Processing Systems 34 (NeurIPS), 2020.
- Gasteiger et al. (2020) Johannes Gasteiger, Janek Groß, and Stephan Günnemann. Directional message passing for molecular graphs. In International Conference on Learning Representations, 2020. URL https://openreview.net/forum?id=B1eWbxStPH.
- Gasteiger et al. (2022) Johannes Gasteiger, Muhammed Shuaibi, Anuroop Sriram, Stephan Günnemann, Zachary Ward Ulissi, C.Lawrence Zitnick, and Abhishek Das. Gemnet-OC: Developing graph neural networks for large and diverse molecular simulation datasets. Transactions on Machine Learning Research, 2022. ISSN 2835-8856. URL https://openreview.net/forum?id=u8tvSxm4Bs.
- Huang et al. (2024) Tinglin Huang, Zhenqiao Song, Rex Ying, and Wengong Jin. Protein-nucleic acid complex modeling with frame averaging transformer. In The Thirty-eighth Annual Conference on Neural Information Processing Systems, 2024. URL https://openreview.net/forum?id=Xngi3Z3wkN.
- Inoue (2018) Hiroshi Inoue. Data augmentation by pairing samples for images classification, 2018. URL https://arxiv.org/abs/1801.02929.
- Jacobs et al. (2025) Ryan Jacobs, Dane Morgan, Siamak Attarian, Jun Meng, Chen Shen, Zhenghao Wu, Clare Yijia Xie, Julia H. Yang, Nongnuch Artrith, Ben Blaiszik, Gerbrand Ceder, Kamal Choudhary, Gabor Csanyi, Ekin Dogus Cubuk, Bowen Deng, Ralf Drautz, Xiang Fu, Jonathan Godwin, Vasant Honavar, Olexandr Isayev, Anders Johansson, Boris Kozinsky, Stefano Martiniani, Shyue Ping Ong, Igor Poltavsky, KJ Schmidt, So Takamoto, Aidan P. Thompson, Julia Westermayr, and Brandon M. Wood. A practical guide to machine learning interatomic potentials – status and future. Current Opinion in Solid State and Materials Science, 35:101214, March 2025. ISSN 1359-0286. doi: 10.1016/j.cossms.2025.101214. URL http://dx.doi.org/10.1016/j.cossms.2025.101214.
- Joshi et al. (2023) Chaitanya K. Joshi, Cristian Bodnar, Simon V Mathis, Taco Cohen, and Pietro Lio. On the expressive power of geometric graph neural networks. In Proceedings of the 40th International Conference on Machine Learning, 2023. URL https://proceedings.mlr.press/v202/joshi23a.html.
- Joshi et al. (2025) Chaitanya K. Joshi, Xiang Fu, Yi-Lun Liao, Vahe Gharakhanyan, Benjamin Kurt Miller, Anuroop Sriram, and Zachary W. Ulissi. All-atom diffusion transformers: Unified generative modelling of molecules and materials. In International Conference on Machine Learning, 2025.
- Kaba et al. (2022) Sékou-Oumar Kaba, Arnab Kumar Mondal, Yan Zhang, Yoshua Bengio, and Siamak Ravanbakhsh. Equivariance with learned canonicalization functions. In NeurIPS 2022 Workshop on Symmetry and Geometry in Neural Representations, 2022. URL https://openreview.net/forum?id=pVD1k8ge25a.
- Klicpera et al. (2021) Johannes Klicpera, Florian Becker, and Stephan Günnemann. Gemnet: Universal directional graph neural networks for molecules. In A.Beygelzimer, Y.Dauphin, P.Liang, and J.Wortman Vaughan (eds.), Advances in Neural Information Processing Systems, 2021. URL https://openreview.net/forum?id=HS_sOaxS9K-.
- Leimeroth et al. (2025) Niklas Leimeroth, Linus C. Erhard, Karsten Albe, and Jochen Rohrer. Machine-learning interatomic potentials from a users perspective: A comparison of accuracy, speed and data efficiency, 2025. URL https://arxiv.org/abs/2505.02503.
- Levine et al. (2025) Daniel S. Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G. Taylor, Muhammad R. Hasyim, Kyle Michel, Ilyes Batatia, Gábor Csányi, Misko Dzamba, Peter Eastman, Nathan C. Frey, Xiang Fu, Vahe Gharakhanyan, Aditi S. Krishnapriyan, Joshua A. Rackers, Sanjeev Raja, Ammar Rizvi, Andrew S. Rosen, Zachary Ulissi, Santiago Vargas, C.Lawrence Zitnick, Samuel M. Blau, and Brandon M. Wood. The open molecules 2025 (omol25) dataset, evaluations, and models, 2025. URL https://arxiv.org/abs/2505.08762.
- Liao & Smidt (2023) Yi-Lun Liao and Tess Smidt. Equiformer: Equivariant graph attention transformer for 3d atomistic graphs. In International Conference on Learning Representations, 2023. URL https://openreview.net/forum?id=KwmPfARgOTD.
- Liao et al. (2024a) Yi-Lun Liao, Brandon Wood, Abhishek Das*, and Tess Smidt*. EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations. In International Conference on Learning Representations (ICLR), 2024a. URL https://openreview.net/forum?id=mCOBKZmrzD.
- Liao et al. (2024b) Yi-Lun Liao et al. Equiformerv2: Improved equivariant transformer for scaling 3d molecular learning. arXiv preprint arXiv:2402.xxxxx, 2024b.
- Lin et al. (2024) Yuchao Lin, Jacob Helwig, Shurui Gui, and Shuiwang Ji. Equivariance via minimal frame averaging for more symmetries and efficiency. In Forty-first International Conference on Machine Learning, 2024. URL https://openreview.net/forum?id=guFsTBXsov.
- Lippmann et al. (2025) Peter Lippmann, Gerrit Gerhartz, Roman Remme, and Fred A Hamprecht. Beyond canonicalization: How tensorial messages improve equivariant message passing. In International Conference on Representation Learning, volume 2025, 2025. URL https://proceedings.iclr.cc/paper_files/paper/2025/file/db7534a06ace69f4ec95bc89e91d5dbb-Paper-Conference.pdf.
- Ma et al. (2024) George Ma, Yifei Wang, Derek Lim, Stefanie Jegelka, and Yisen Wang. A canonicalization perspective on invariant and equivariant learning. In The Thirty-eighth Annual Conference on Neural Information Processing Systems, 2024. URL https://openreview.net/forum?id=jjcY92FX4R.
- Maruf et al. (2025) Moin Uddin Maruf, Sungmin Kim, and Zeeshan Ahmad. Learning long-range interactions in equivariant machine learning interatomic potentials via electronic degrees of freedom. The Journal of Physical Chemistry Letters, 16(35):9078–9087, August 2025. ISSN 1948-7185. doi: 10.1021/acs.jpclett.5c02352. URL http://dx.doi.org/10.1021/acs.jpclett.5c02352.
- Mazitov et al. (2025) Arslan Mazitov, Filippo Bigi, Matthias Kellner, Paolo Pegolo, Davide Tisi, Guillaume Fraux, Sergey Pozdnyakov, Philip Loche, and Michele Ceriotti. Pet-mad, a lightweight universal interatomic potential for advanced materials modeling, 2025. URL https://arxiv.org/abs/2503.14118.
- Musaelian et al. (2023) Albert Musaelian, Sebastian Batzner, Anders Johansson, Simon Kozinsky, and Boris Kozinsky. Learning local 3d energetics with graph neural networks. Nature Communications, 2023.
- Negassi et al. (2022) Misgana Negassi, Diane Wagner, and Alexander Reiterer. Smart(sampling)augment: Optimal and efficient data augmentation for semantic segmentation. Algorithms, 15(5), 2022. ISSN 1999-4893. doi: 10.3390/a15050165. URL https://www.mdpi.com/1999-4893/15/5/165.
- Noé et al. (2020) Frank Noé, Alexandre Tkatchenko, Klaus-Robert Müller, and Cecilia Clementi. Machine learning for molecular simulation. Annual Review of Physical Chemistry, 71(1):361–390, April 2020. ISSN 1545-1593. doi: 10.1146/annurev-physchem-042018-052331. URL http://dx.doi.org/10.1146/annurev-physchem-042018-052331.
- Passaro & Zitnick (2023a) Saro Passaro and C.Lawrence Zitnick. Reducing so(3) convolutions to so(2) for efficient equivariant gnns. In Proceedings of the 40th International Conference on Machine Learning, ICML’23. JMLR.org, 2023a.
- Passaro & Zitnick (2023b) Saro Passaro and C.Lawrence Zitnick. Reducing SO(3) convolutions to SO(2) for efficient equivariant GNNs. In Proceedings of the 40th International Conference on Machine Learning, Proceedings of Machine Learning Research. PMLR, 2023b.
- Power et al. (2022) Alethea Power, Yuri Burda, Harri Edwards, Igor Babuschkin, and Vedant Misra. Grokking: Generalization beyond overfitting on small algorithmic datasets. arXiv preprint arXiv:2201.02177, 2022.
- Puny et al. (2022) Omri Puny, Matan Atzmon, Edward J. Smith, Ishan Misra, Aditya Grover, Heli Ben-Hamu, and Yaron Lipman. Frame averaging for invariant and equivariant network design. In International Conference on Learning Representations, 2022. URL https://openreview.net/forum?id=zIUyj55nXR.
- Qu & Krishnapriyan (2024) Eric Qu and Aditi Krishnapriyan. The importance of being scalable: Improving the speed and accuracy of neural network interatomic potentials across chemical domains. Advances in Neural Information Processing Systems, 37:139030–139053, 2024.
- Rahat et al. (2024) Fazle Rahat, M Shifat Hossain, Md Rubel Ahmed, Sumit Kumar Jha, and Rickard Ewetz. Data augmentation for image classification using generative ai, 2024. URL https://arxiv.org/abs/2409.00547.
- Rhodes et al. (2025) Benjamin Rhodes, Sander Vandenhaute, Vaidotas Šimkus, James Gin, Jonathan Godwin, Tim Duignan, and Mark Neumann. Orb-v3: atomistic simulation at scale, 2025. URL https://arxiv.org/abs/2504.06231.
- Sanchez-Gonzalez et al. (2020) Alvaro Sanchez-Gonzalez, Jonathan Godwin, Tobias Pfaff, Rex Ying, Jure Leskovec, and Peter W. Battaglia. Learning to simulate complex physics with graph networks. In Proceedings of the 37th International Conference on Machine Learning. JMLR.org, 2020.
- Satorras et al. (2021) Victor Garcia Satorras, Emiel Hoogeboom, and Max Welling. E(n) equivariant graph neural networks. In Proceedings of the 38rd International Conference on Machine Learning, 2021.
- Schütt et al. (2017) Kristof T. Schütt, Huziel E. Sauceda, P.-J. Kindermans, Alexandre Tkatchenko, and Klaus-Robert Müller. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. In NeurIPS, 2017.
- Shuaibi et al. (2025) Muhammed Shuaibi, Abhishek Das, Anuroop Sriram, Misko, Luis Barroso-Luque, Ray Gao, Siddharth Goyal, Zachary Ulissi, Brandon Wood, Tian Xie, Junwoong Yoon, Brook Wander, Adeesh Kolluru, Richard Barnes, Ethan Sunshine, Kevin Tran, Xiang, Daniel Levine, Nima Shoghi, Ilias Chair, , Janice Lan, Kaylee Tian, Joseph Musielewicz, clz55, Weihua Hu, , Kyle Michel, willis, and vbttchr. FAIRChem, 2025. URL https://github.com/facebookresearch/fairchem.
- Su et al. (2023) Jianlin Su, Yu Lu, Shengfeng Pan, Ahmed Murtadha, Bo Wen, and Yunfeng Liu. Roformer: Enhanced transformer with rotary position embedding, 2023. URL https://arxiv.org/abs/2104.09864.
- Thölke & Fabritiis (2022) Philipp Thölke and Gianni De Fabritiis. Equivariant transformers for neural network based molecular potentials. In International Conference on Learning Representations, 2022. URL https://openreview.net/forum?id=zNHzqZ9wrRB.
- Wang et al. (2024) Yuyang Wang, Ahmed A. Elhag, Navdeep Jaitly, Joshua M. Susskind, and Miguel Ángel Bautista. Swallowing the bitter pill: Simplified scalable conformer generation. In Forty-first International Conference on Machine Learning, 2024.
- Yang et al. (2025) Ziduo Yang, Xian Wang, Yifan Li, Qiujie Lv, Calvin Yu-Chian Chen, and Lei Shen. Efficient equivariant model for machine learning interatomic potentials. npj Computational Materials, 11(1):49, 2025. doi: 10.1038/s41524-025-01535-3. URL https://doi.org/10.1038/s41524-025-01535-3.
- Yu et al. (2023) Xinyi Yu, Guanbin Li, Wei Lou, Siqi Liu, Xiang Wan, Yan Chen, and Haofeng Li. Diffusion-based data augmentation for nuclei image segmentation. In Medical Image Computing and Computer Assisted Intervention – MICCAI 2023, 2023.
- Yuan et al. (2025) Eric C-Y Yuan, Yunsheng Liu, Junmin Chen, Peichen Zhong, Sanjeev Raja, Tobias Kreiman, Santiago Vargas, Wenbin Xu, Martin Head-Gordon, Chao Yang, et al. Foundation models for atomistic simulation of chemistry and materials. arXiv preprint arXiv:2503.10538, 2025.
- Zhang et al. (2025) Leo Zhang, Kianoosh Ashouritaklimi, Yee Whye Teh, and Rob Cornish. Symdiff: Equivariant diffusion via stochastic symmetrisation. In The Thirteenth International Conference on Learning Representations, 2025. URL https://openreview.net/forum?id=i1NNCrRxdM.
- Zhang et al. (2018) Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, and Weinan E. Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics. Physical Review Letters, 120(14), April 2018. ISSN 1079-7114. doi: 10.1103/physrevlett.120.143001. URL http://dx.doi.org/10.1103/PhysRevLett.120.143001.
Appendix A Implementation Details
A.1 Model Architecture
Table2 provides the complete architectural specifications for TransIP’s model versions.
Table 2: TransIP model configurations. All versions share the same embedding method and activation functions.
A.2 Training Hyperparameters
Table 3 provides TransIP’s optimal hyperparameters.
Table 3: Training hyperparameters used for all TransIP experiments.
A.3 Data Processing and Augmentation
TransIP processes molecular data with the following pipeline:
- •Coordinate centering: Atomic coordinates are centered by subtracting the center of mass: 𝐫 i←𝐫 i−1|m|∑j 𝐫 j\mathbf{r}{i}\leftarrow\mathbf{r}{i}-\frac{1}{|m|}\sum_{j}\mathbf{r}_{j}
- •Equivariance pairs: For training with learned equivariance, we create pairs (m,ϕ(g)(m))(m,\phi(g)(m)) where g g is sampled uniformly from SO(3)\mathrm{SO(3)} per molecule.
A.4 Evaluation Metrics
We evaluate model performance using the following metrics:
Force Mean Absolute Error (MAE):
Force MAE=1 3|m|∑i=1 N∑α∈{x,y,z}|𝐅 i,α−𝐅 i,α∗|(eV/Å)\text{Force MAE}=\frac{1}{3|m|}\sum_{i=1}^{N}\sum_{\alpha\in{x,y,z}}|\mathbf{F}{i,\alpha}-\mathbf{F}^{*}{i,\alpha}|\quad\text{(eV/\AA )}(8)
Force Cosine Similarity:
Force CosSim=1|m|∑i=1|m|𝐅 i⋅𝐅 i∗‖𝐅 i‖‖𝐅 i∗‖\text{Force CosSim}=\frac{1}{|m|}\sum_{i=1}^{|m|}\frac{\mathbf{F}{i}\cdot\mathbf{F}^{*}{i}}{|\mathbf{F}{i}||\mathbf{F}^{*}{i}|}(9)
Energy per Atom MAE:
Energy/atom MAE=1|m||E−E∗|(eV/atom)\text{Energy/atom MAE}=\frac{1}{|m|}|E-E^{*}|\quad\text{(eV/atom)}(10)
Total Energy MAE:
Total Energy MAE=|E−E∗|(eV)\text{Total Energy MAE}=|E-E^{*}|\quad\text{(eV)}(11)
where 𝐅\mathbf{F} and E E denote predicted forces and energies, 𝐅∗\mathbf{F}^{} and E∗E^{} are ground truth values, and |m||m| is the total number of atoms. For energies, we use referenced targets following Levine et al. (2025).
A.5 Computational Resources
- •5-epoch experiments: 8 NVIDIA 80GB GPUs
- •80-epoch experiments: 64 NVIDIA 80GB GPUs
A.6 Validation Splits
For 5-epoch runs, we evaluate on domain-specific validation subsets sampled from the OMol25 validation (Val-Comp) dataset:
- •Metal complexes: 20,000 samples
- •Electrolytes: 20,000 samples
- •Biomolecules: 20,000 samples
- •SPICE: 9,630 samples (complete subset)
- •Neutral organics: 20,000 samples (including ANI2x, OrbNet-Denali, GEOM, Trans1x, RGD)
- •Reactivity: 20,000 samples
- •Full validation set: 20,000 samples.
We use the full (2M) Val Comp dataset to evaluate TransIP and TransAug in Table 1.
Appendix B Additional Results
In this section, we include additional dataset scaling results for TransIP and TransAug.
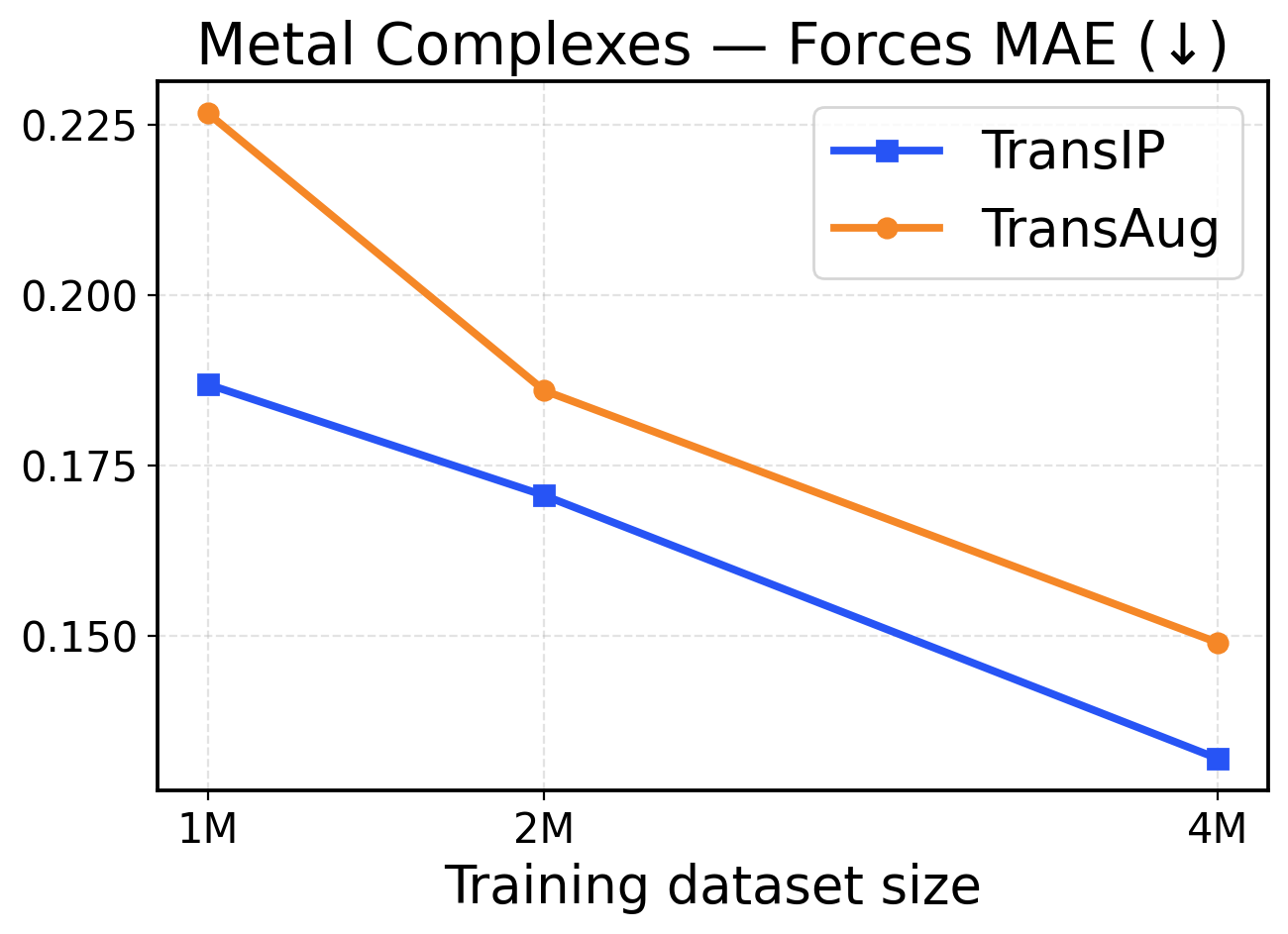

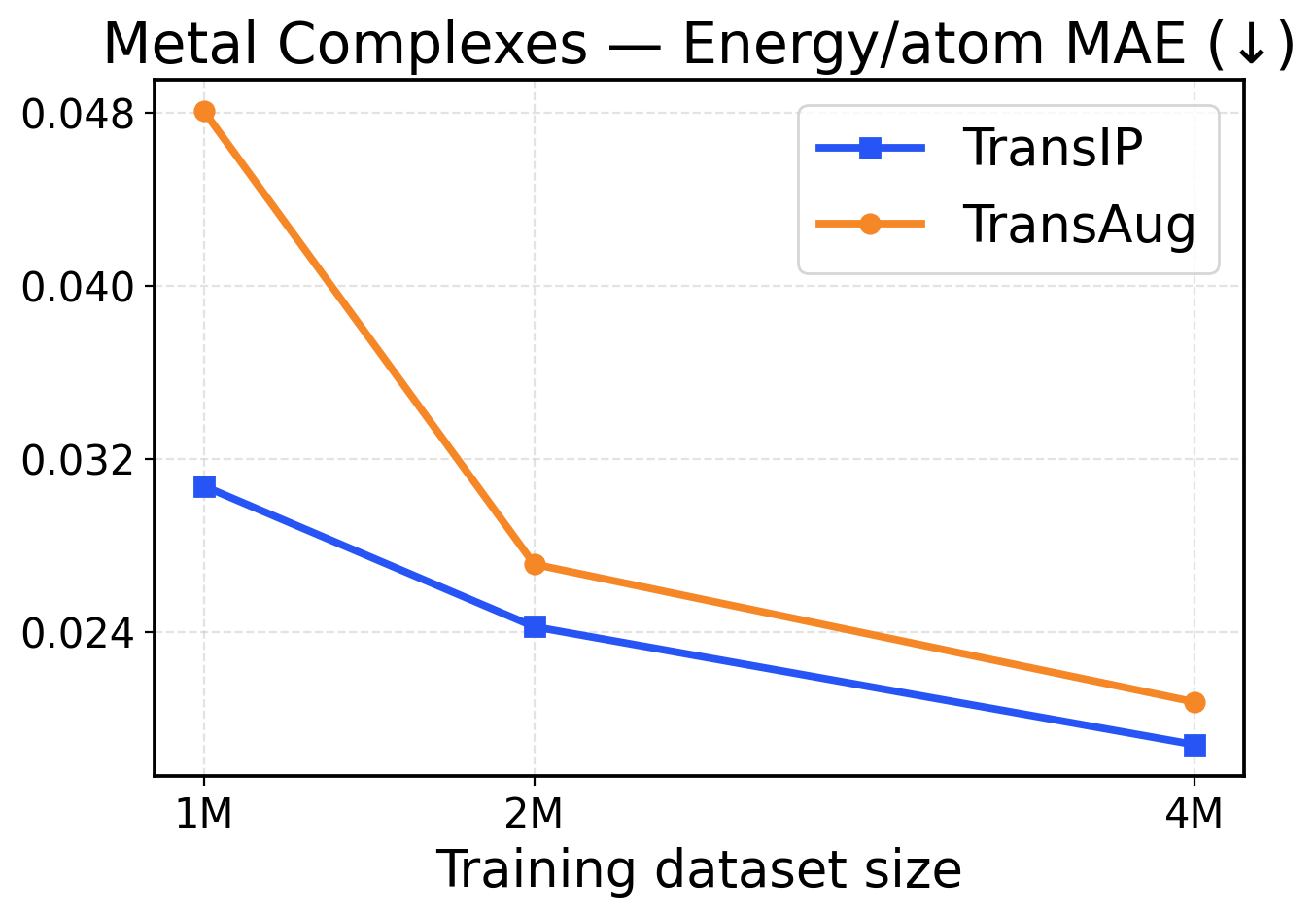
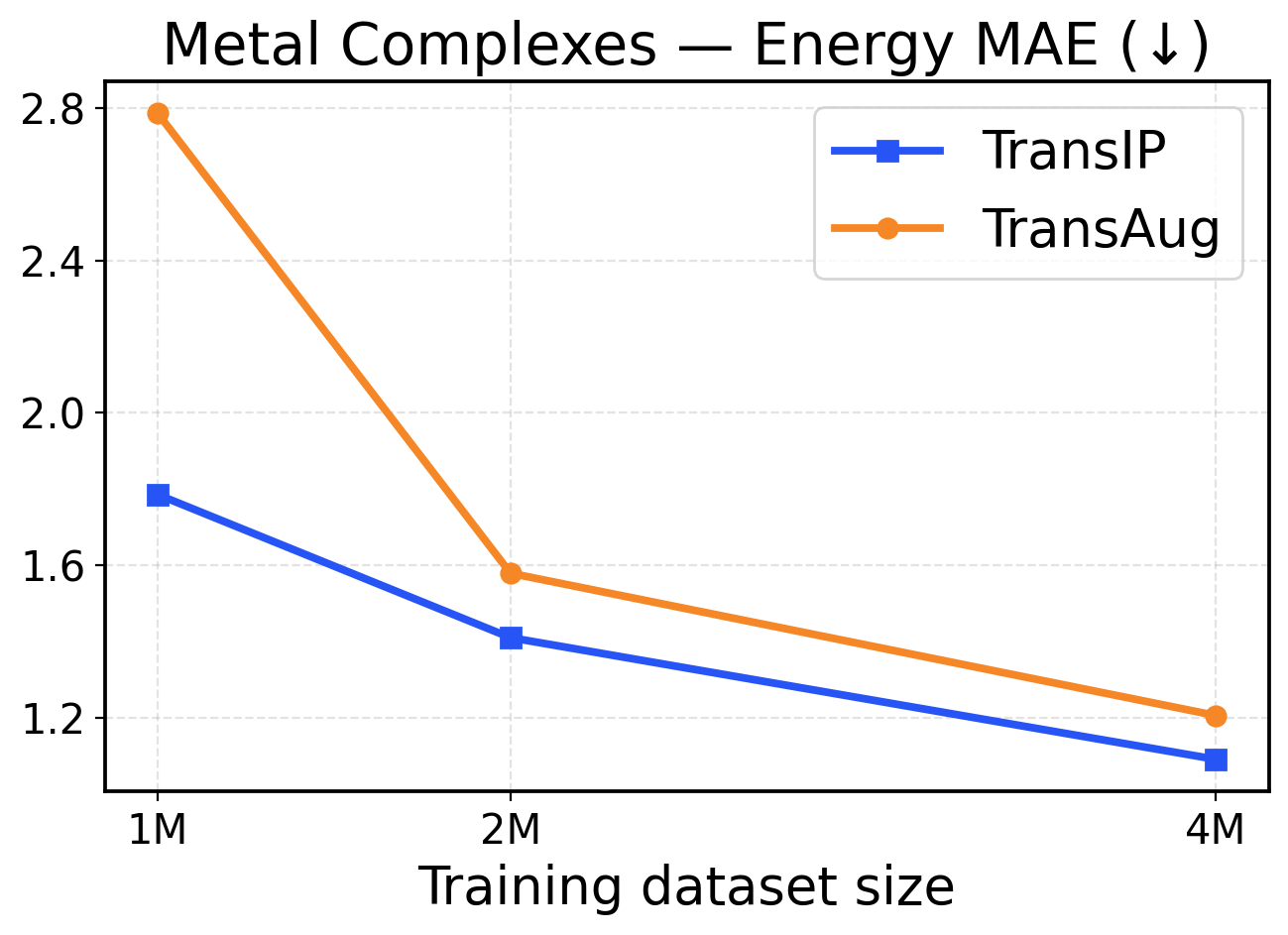
Figure 5: Metal Complexes scaling across training dataset sizes (1M / 2M / 4M). The top row presents force metrics, while the bottom row displays energy metrics.
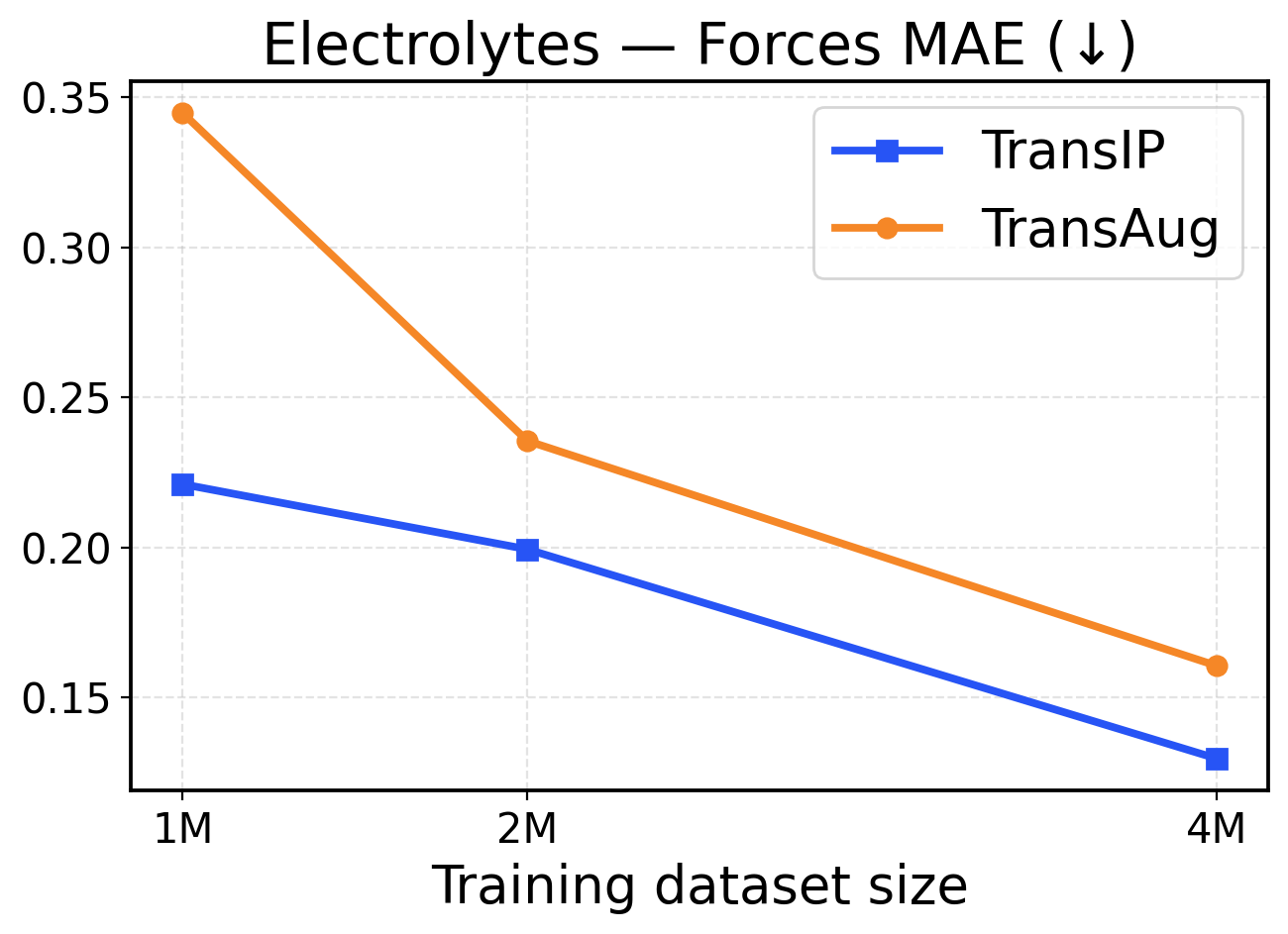



Figure 6: Electrolytes scaling across training dataset sizes (1M / 2M / 4M). The top row presents force metrics, while the bottom row displays energy metrics.
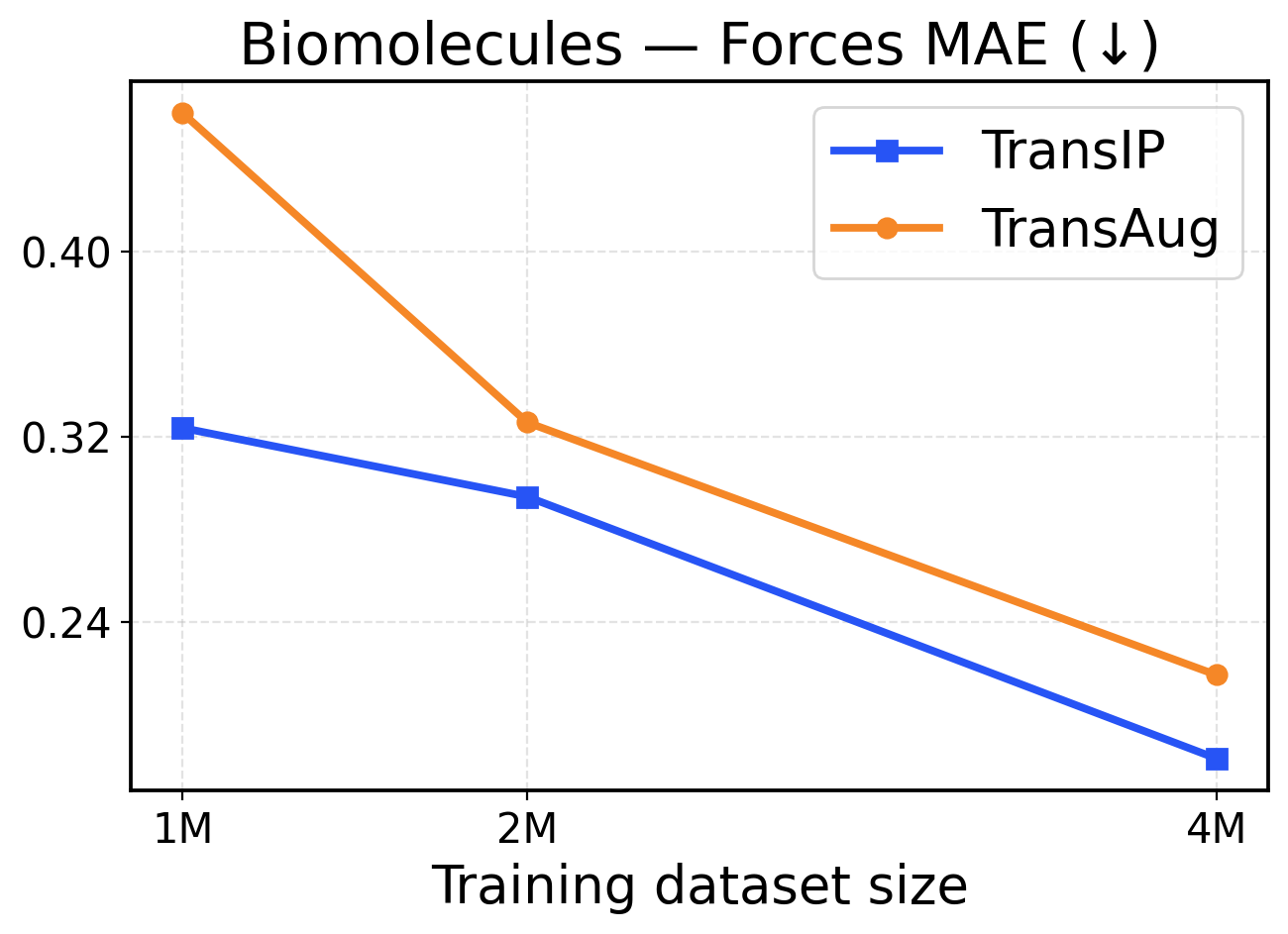

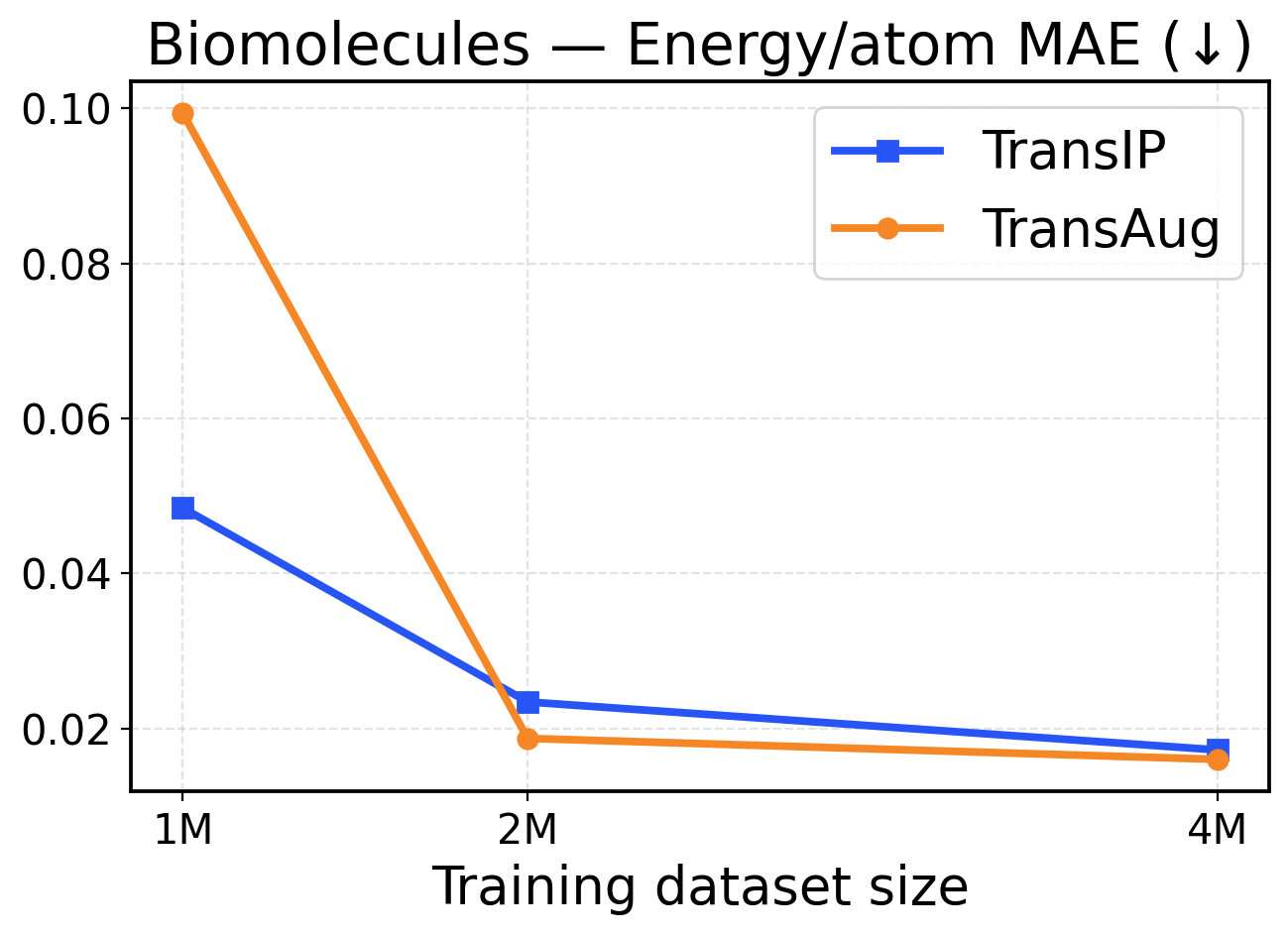

Figure 7: Biomolecules scaling across training dataset sizes (1M / 2M / 4M). The top row presents force metrics, while the bottom row displays energy metrics.
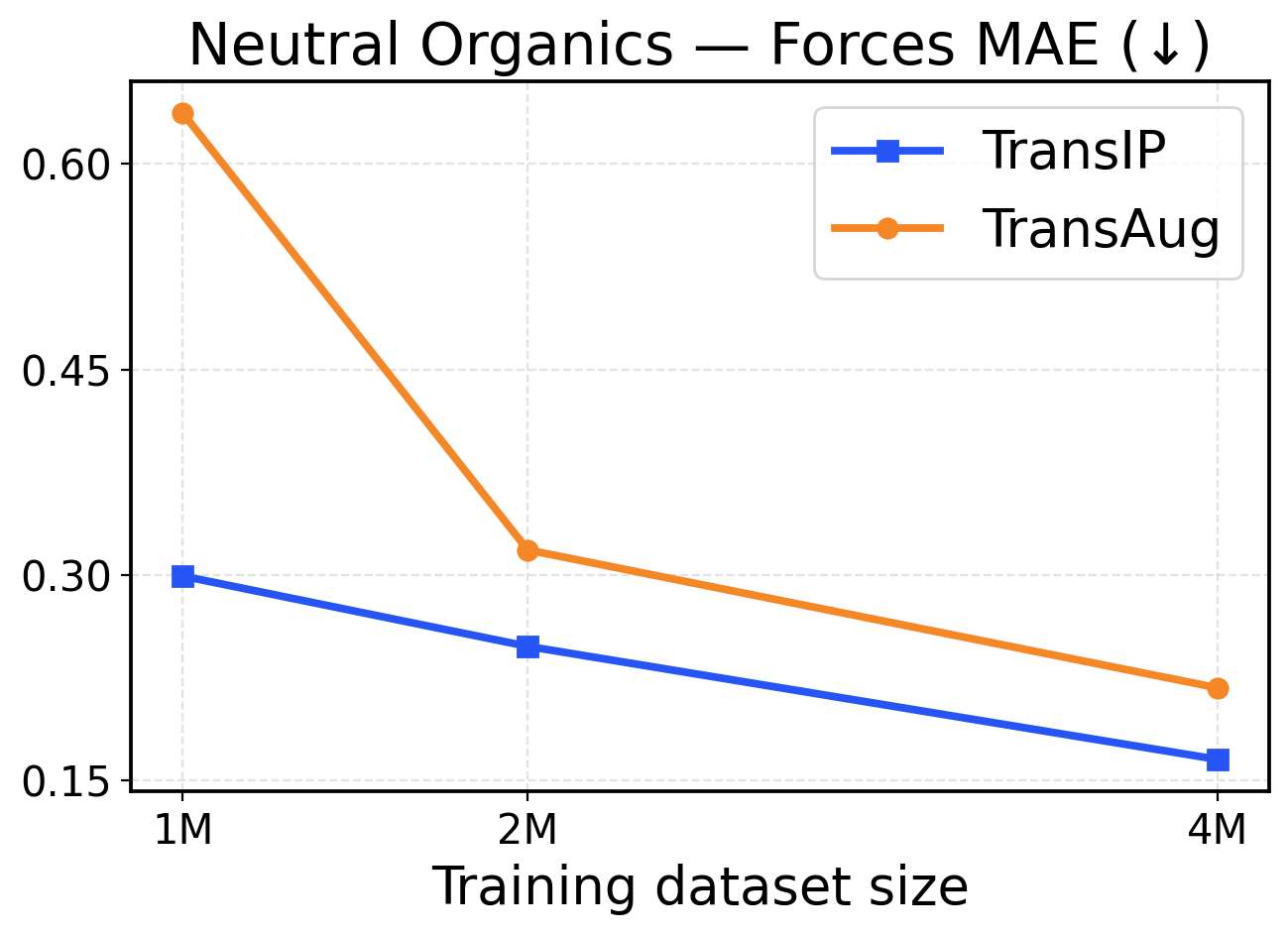

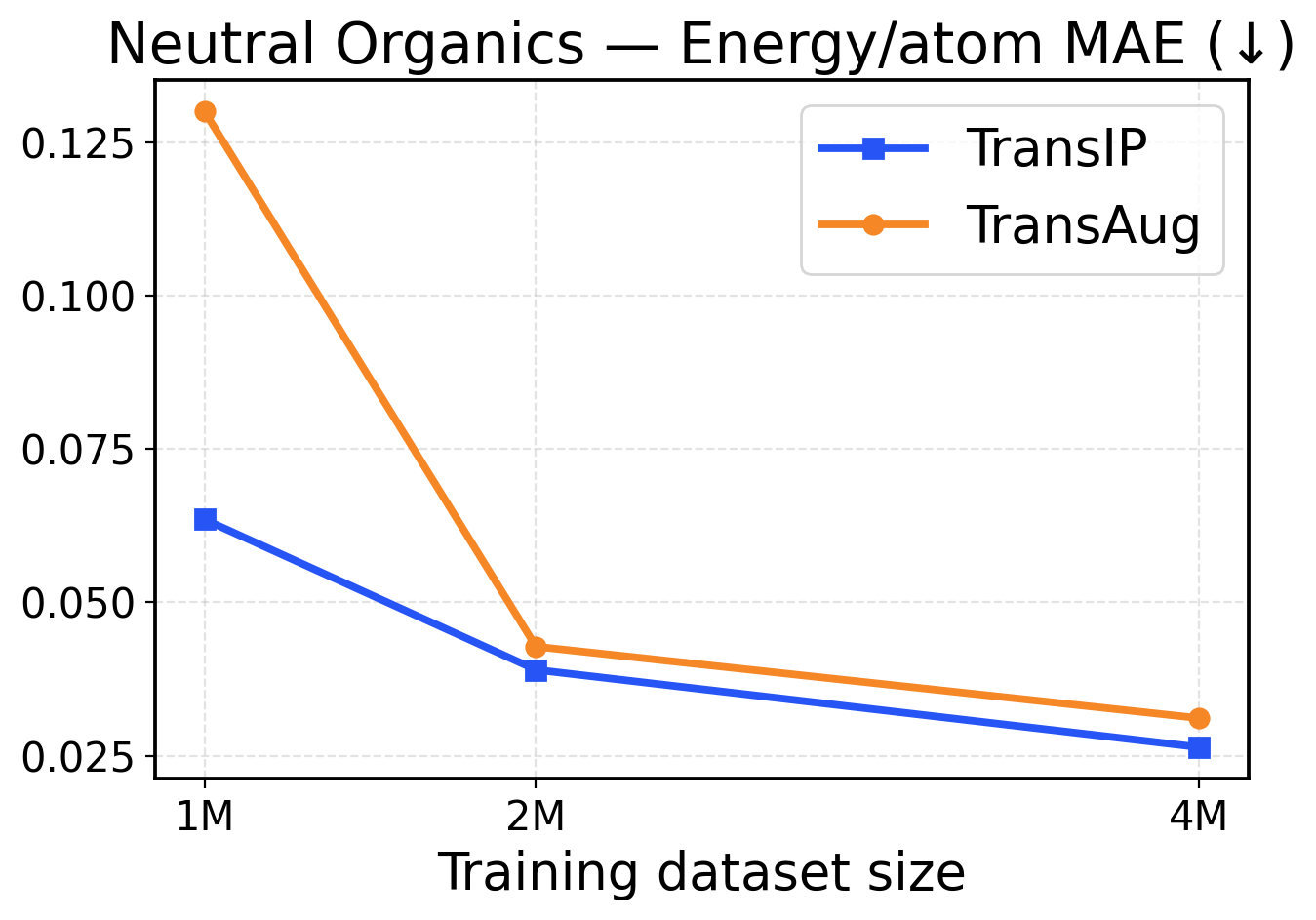

Figure 8: Neutral Organics scaling across training dataset sizes (1M / 2M / 4M). The top row presents force metrics, while the bottom row displays energy metrics.
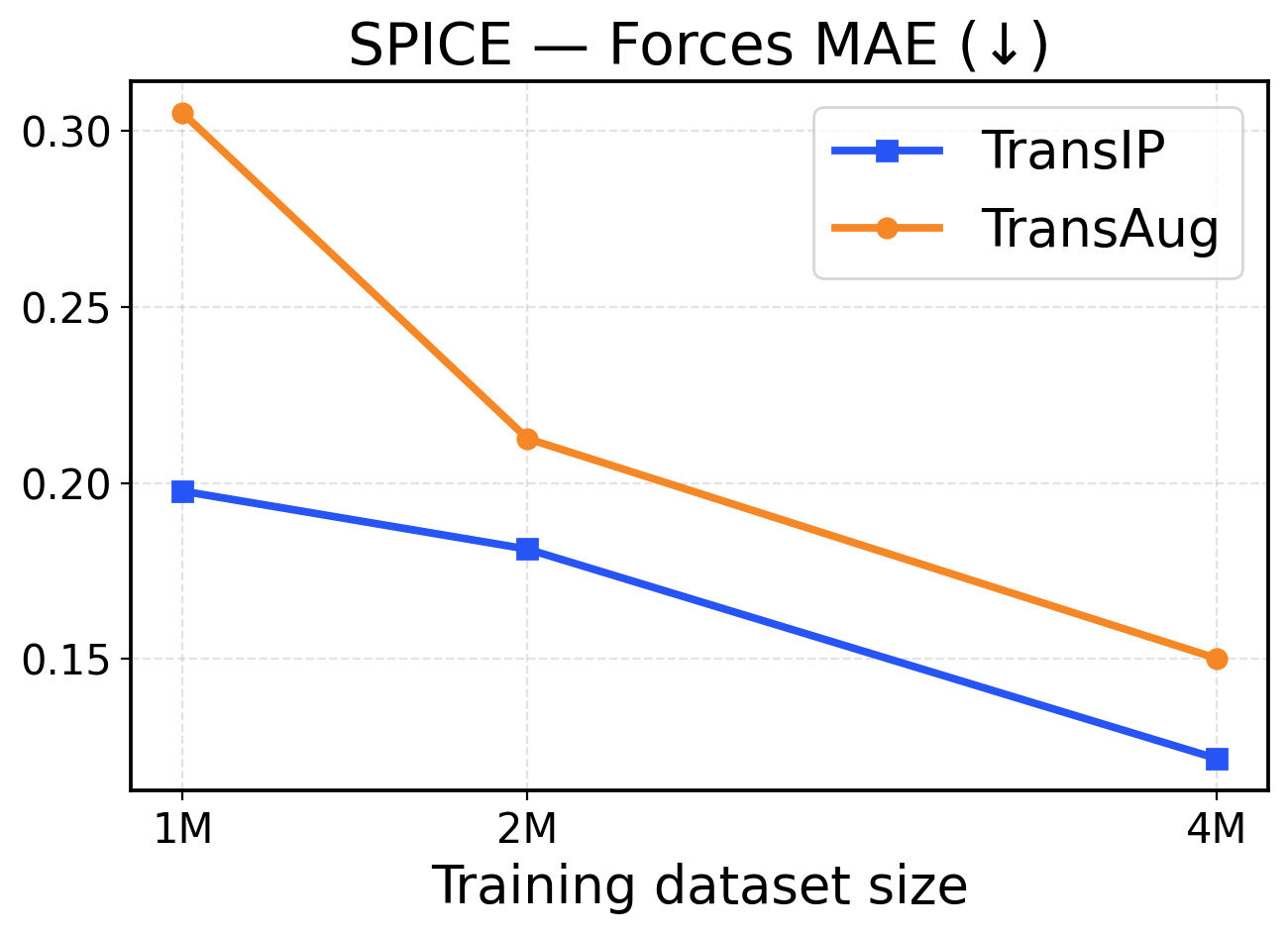

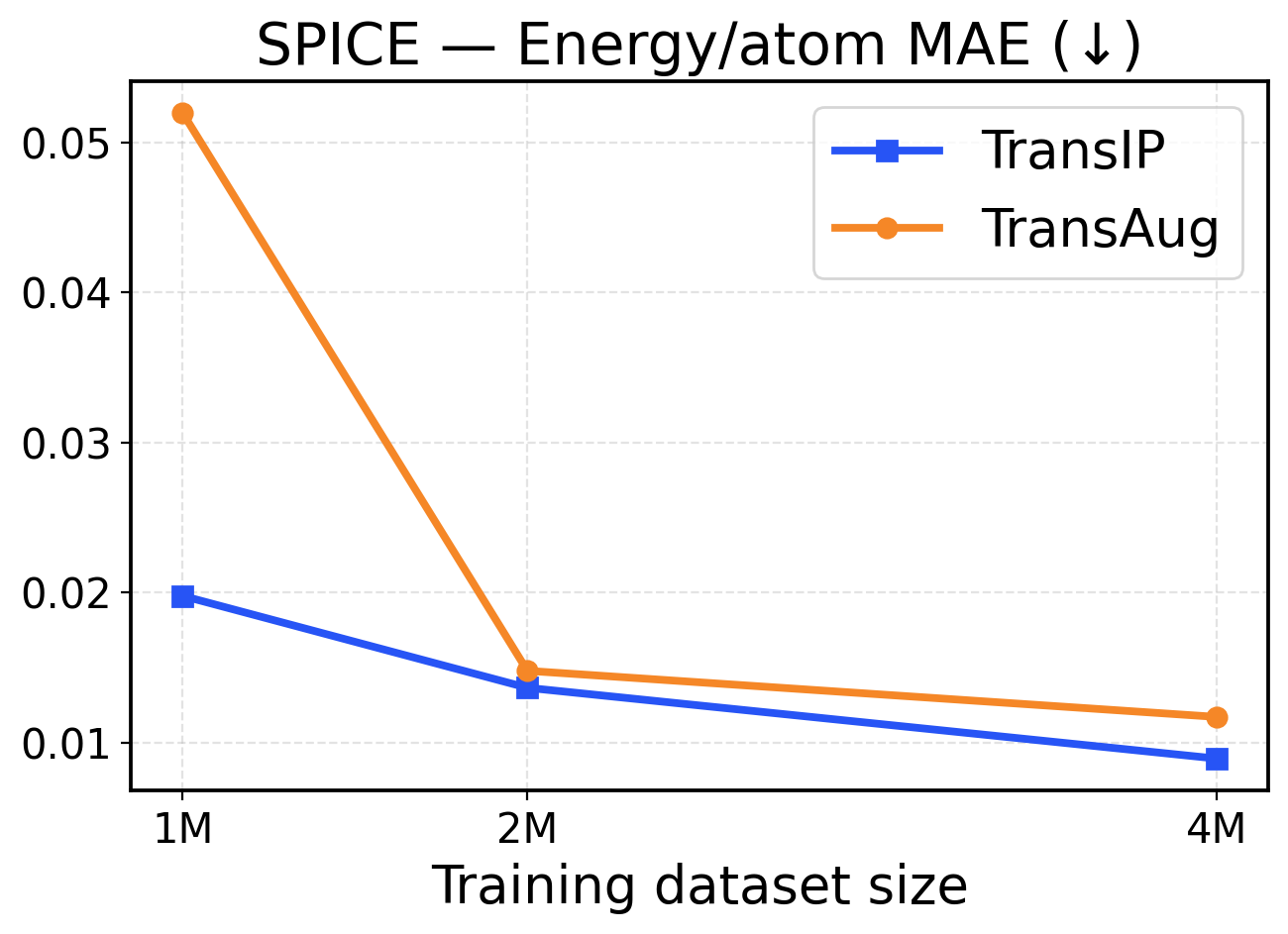
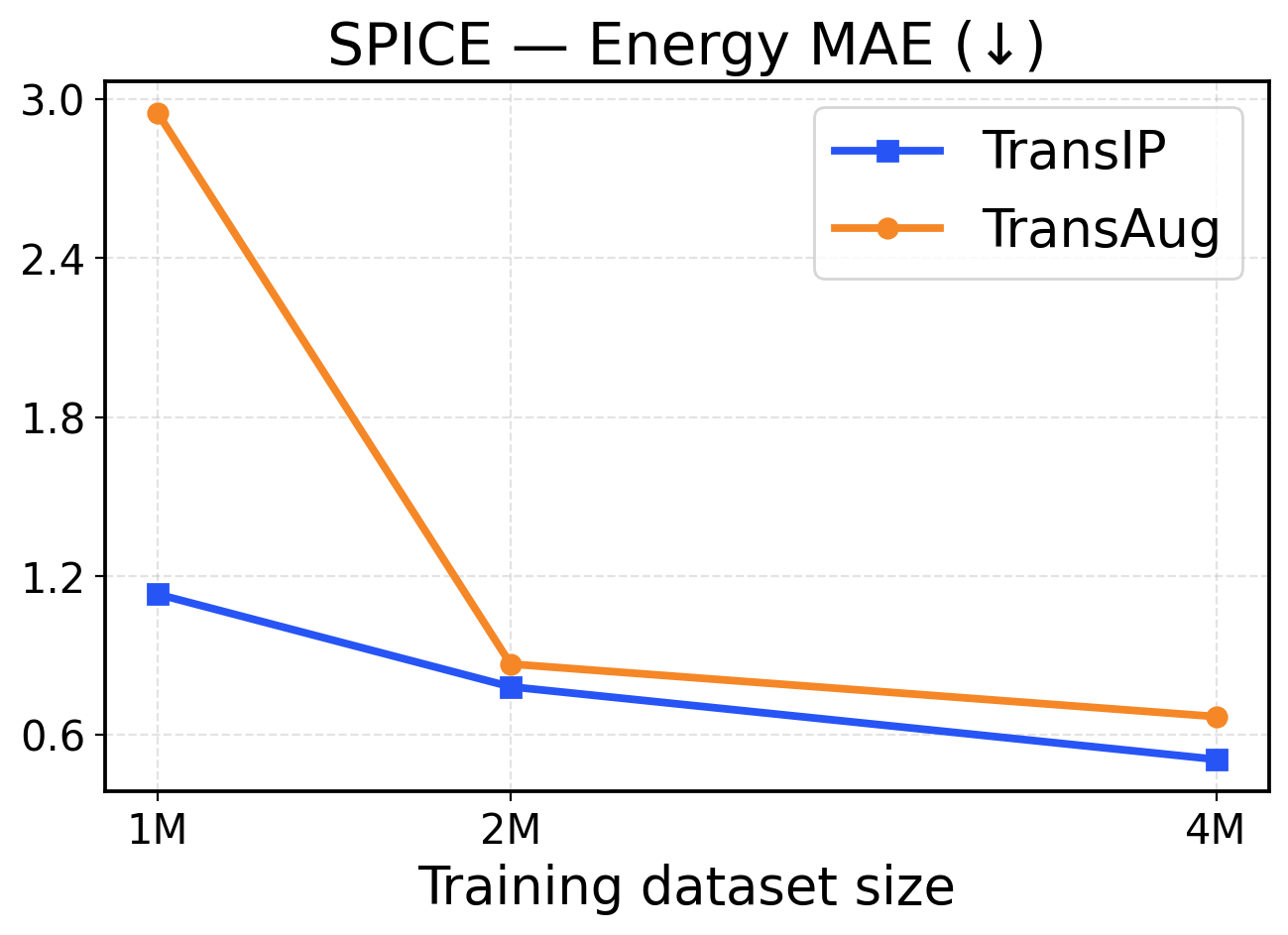
Figure 9: SPICE scaling across training dataset sizes (1M / 2M / 4M). The top row presents force metrics, while the bottom row displays energy metrics.
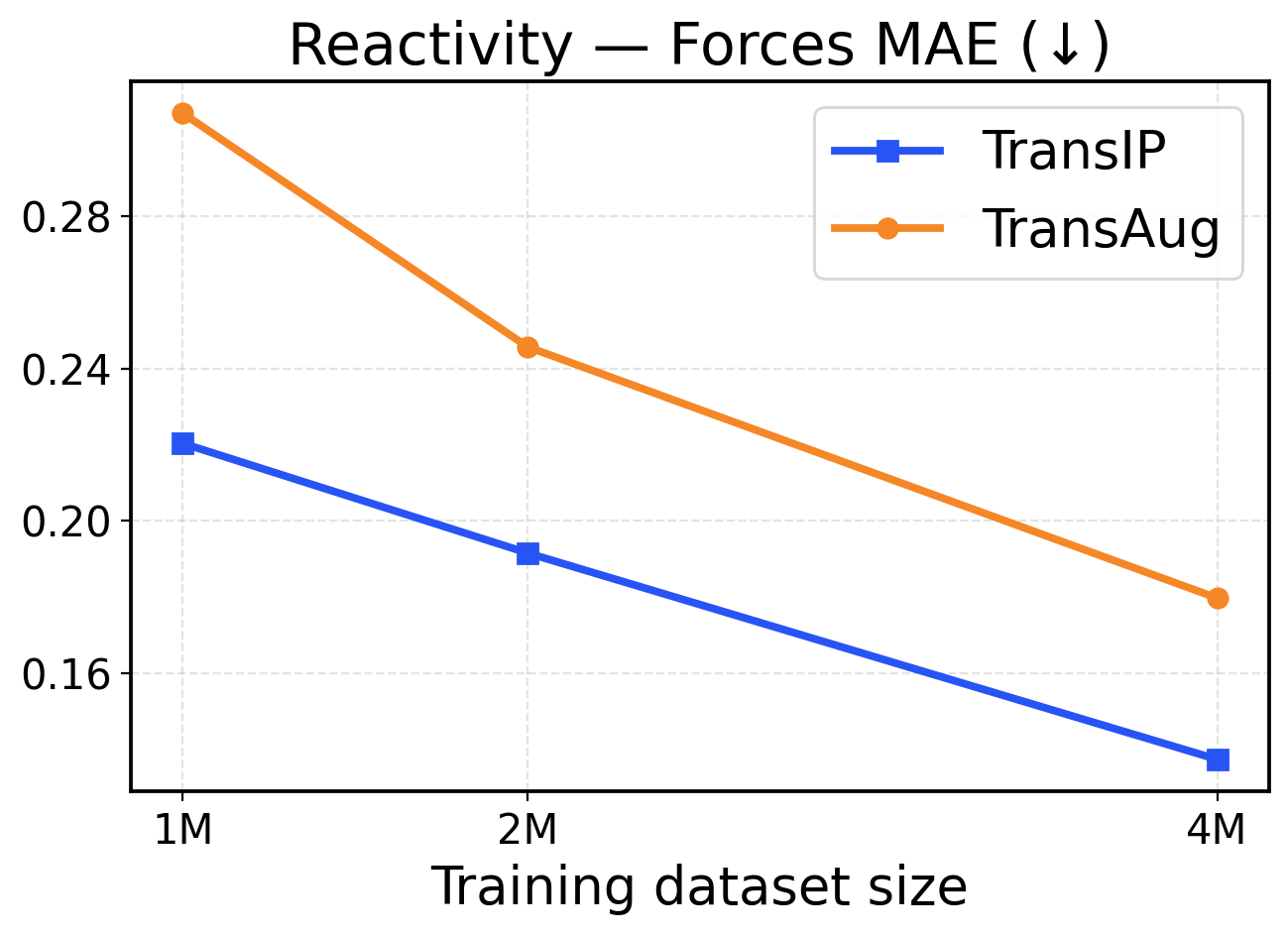

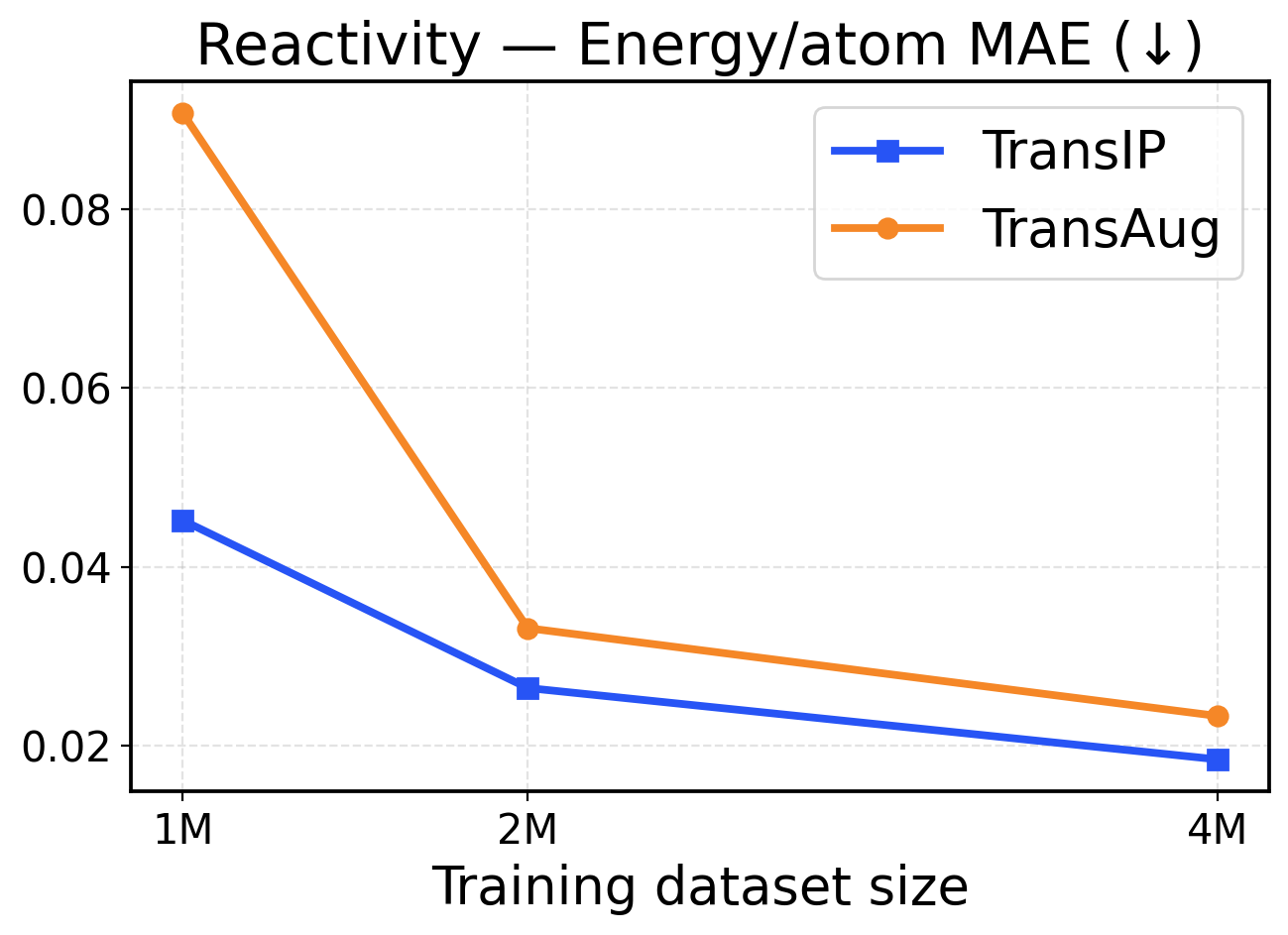
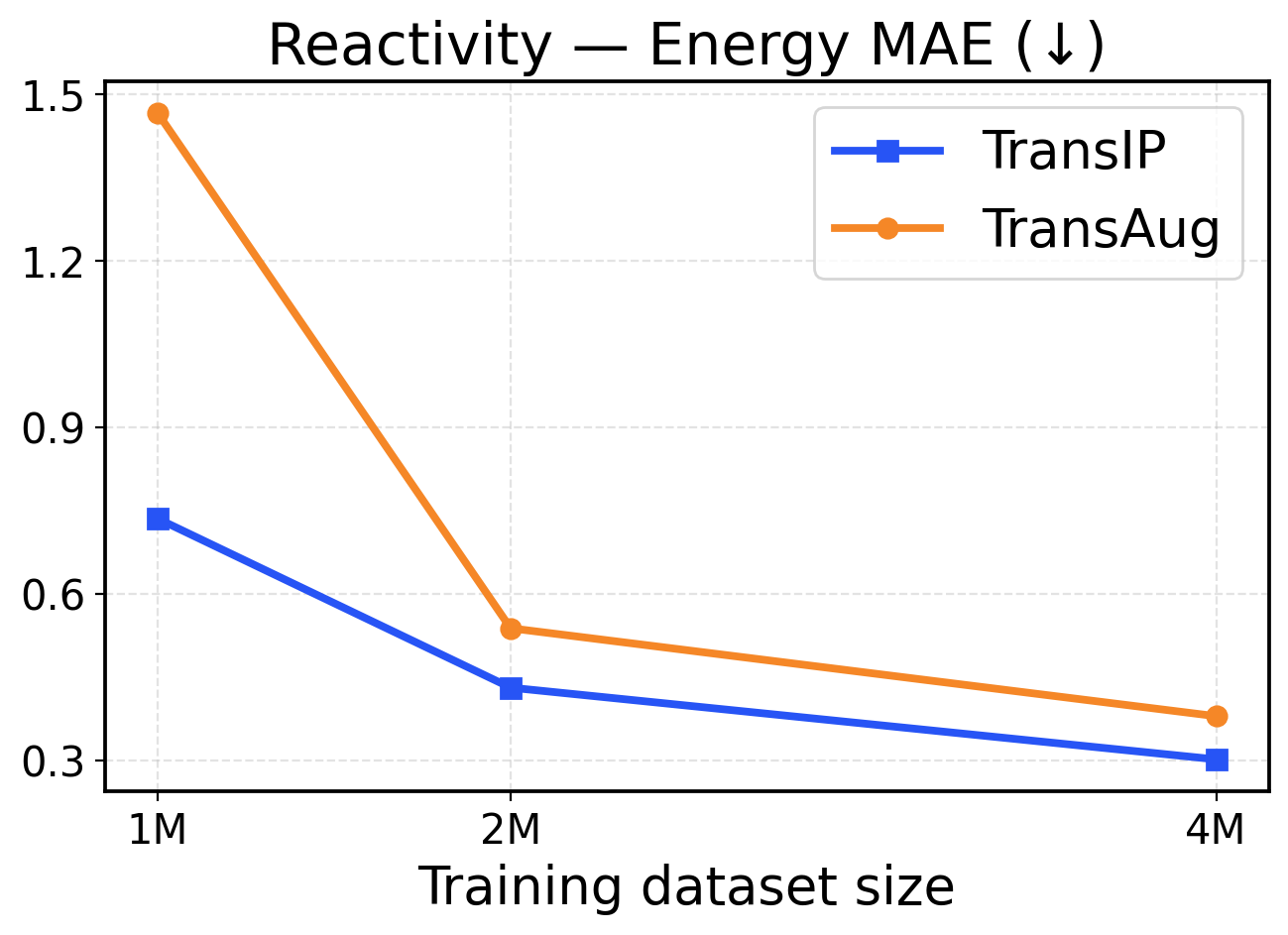
Figure 10: Reactivity scaling across training dataset sizes (1M / 2M / 4M). The top row presents force metrics, while the bottom row displays energy metrics.
Xet Storage Details
- Size:
- 64.2 kB
- Xet hash:
- 03d25ad2d99cd84f3c67a62c94729d75b58a62a5143482258dcbd870c6274117
Xet efficiently stores files, intelligently splitting them into unique chunks and accelerating uploads and downloads. More info.